Integrating multiple modalities for precise cell heterogeneity identification
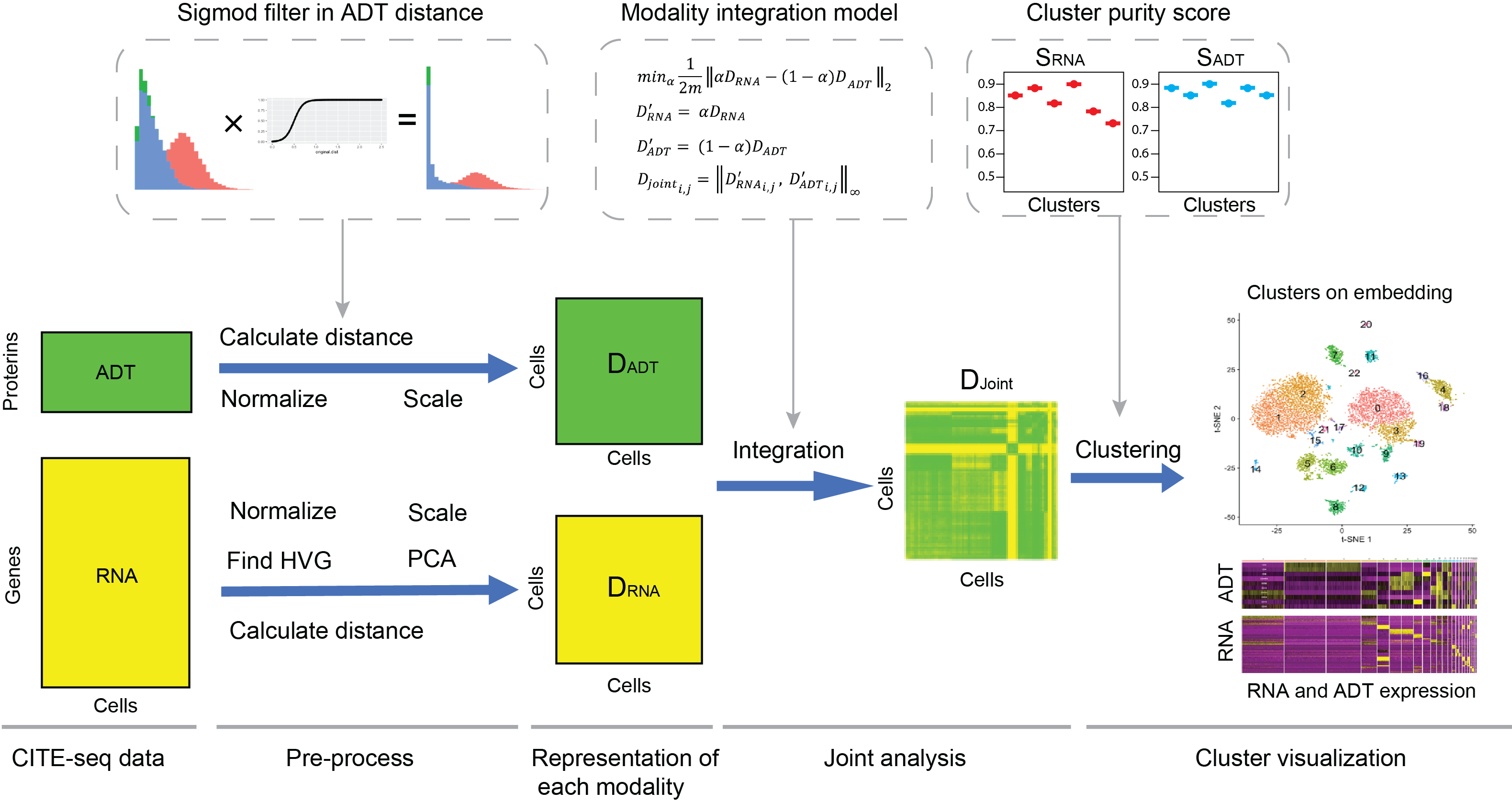
“LinQ-View” was designed as a joint single cell analysis strategy that could integrate information from both transcriptome and surface protein markers for cell heterogeneity identification. The system structure was inspried by Seurat, PAGA and other conventional scRNA-seq tools. We propose quantitive metric for cluster purity of CITE-seq data, enabling effective determination of clustering algorithms and their parameters. We demonstrate the utility of our toolkit through seamless integration with standard single cell analysis workflows on several public datasets. Through comparison to existing multimodal methods, we demonstrate that LinQ-View generates more accurate cell clusters and is specialized in handling of CITE-seq data with limited ADT features (e.g. less than 50). Furthermore, we also demonstrate the potential of LinQ-View on integrating antigen probe binding with transcriptome expression to identify a unique phenotype: antigen specificity of B cells. Finally, we analyze single cell transcriptional and protein expression from COVID-19-infected patients and influenza-immunized subjects, revealing antigen-specific B cell subsets and previously unknown T cell subsets post-infection and vaccination.
We use cell-cell distances to represent variations of gene expression in each modality. Cell-cell distances from different modalities could be scaled into same level by a linear transformation, and then being integrated into one distance matrix, which will be able to represent variations of gene expression from multiple modalities. We introduced L-infinite norm model for the distance integration. Since the variations among cells were represented by a cell-cell distance matrix, “LinQ-View” is compatible with all clustering methods (e.g. k-means, Hierarchical clustering, community detection, Louvain, FCM) and dimension reduction methods (e.g. MDS, t-SNE, UMAP). After cell embedding and clustering were constructed, modalities in genotype/phenotype property group could be applied to the result for biological patterns identification and enrichment analysis. We believe that this method could help to identify cell heterogeneity more accurately and also benefit further downstream analysis.
For more details about our model, please refer to our paper: #Li, Lei; #Dugan, Haley L; #Stamper, Christopher et al. Improved integration of single-cell transcriptome and surface protein expression by LinQ-View Cell Reports Methods 1 (4), 100056 (2021)
Highlights of LinQ-View
- We proposed a quantitive metric for selection of clustering methods and their parameters
- Compare to existing methods, LinQ-View is specialized in handling CITE-seq data with limited ADT features
- LinQ-View is able to reveal unique phenotype (antigen-specificity) of immune cells