User manual too long? Try asking this AI notebook
VGenes Notebook LLMGet started
- Create a new BCR/TCR database or open our example database
- Import BCR/TCR sequences into your database
- Then enjoy VGenes to analyze and clone your sequences
Data import and annotation
-
Create VGenes database
On the welcome page, users are allowed to create new a VGenes database (DB) or open an existing VGenes DB. Users are allowed to check “backup my DB” to make a backup for the database being opened. This backup function is disabled by default for faster response. All recently visited VGenes DBs will be saved in a list, and users are allowed to quickly visit them using the “open recent” dropdown combo box.
-
Import data
New users can click “New VGene DB” to create their first VGenes database. After the VGenes DB is created, the program will ask users to import data into this database. Users will be directed to a data import dialog after they click “Yes”. Notably, users still have chance to import data into their database if they accidently clicked “No”. Users can click "data import icon" on the main interface to open the data import dialog.
As shown in the following figure, VGenes allows users to import raw data from multiple sources: 1) 10X single cell VDJ profiling generated by CellRanger, 2) FASTA files, 3) SEQ files. They are most commonly used data formats for either single-cell or bulk sequencing data in the field. VGenes supports both B cell receptor (BCR) and T cell receptor (TCR) data. Users can click the BCR and TCR icons to switch between two data types.
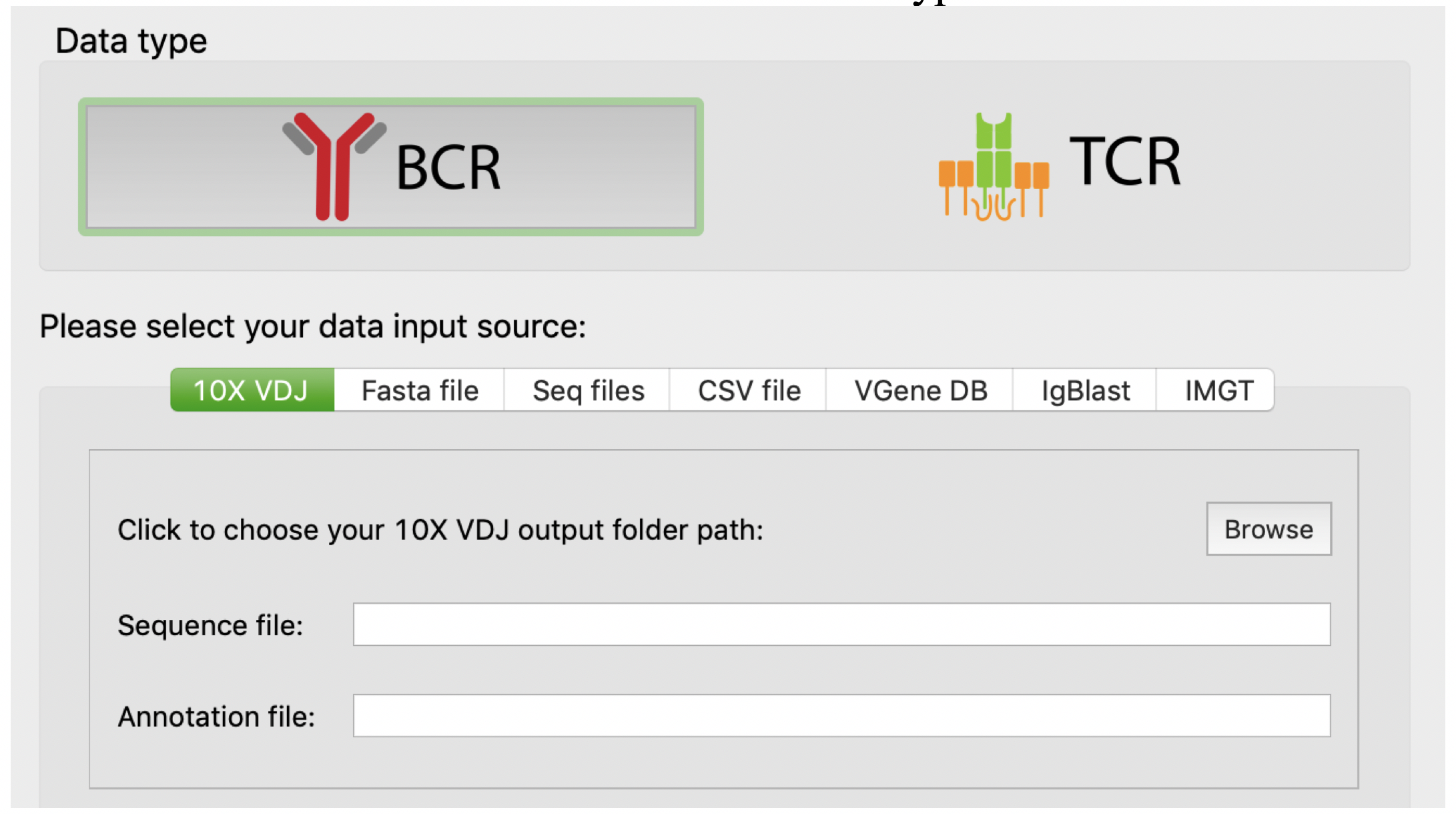
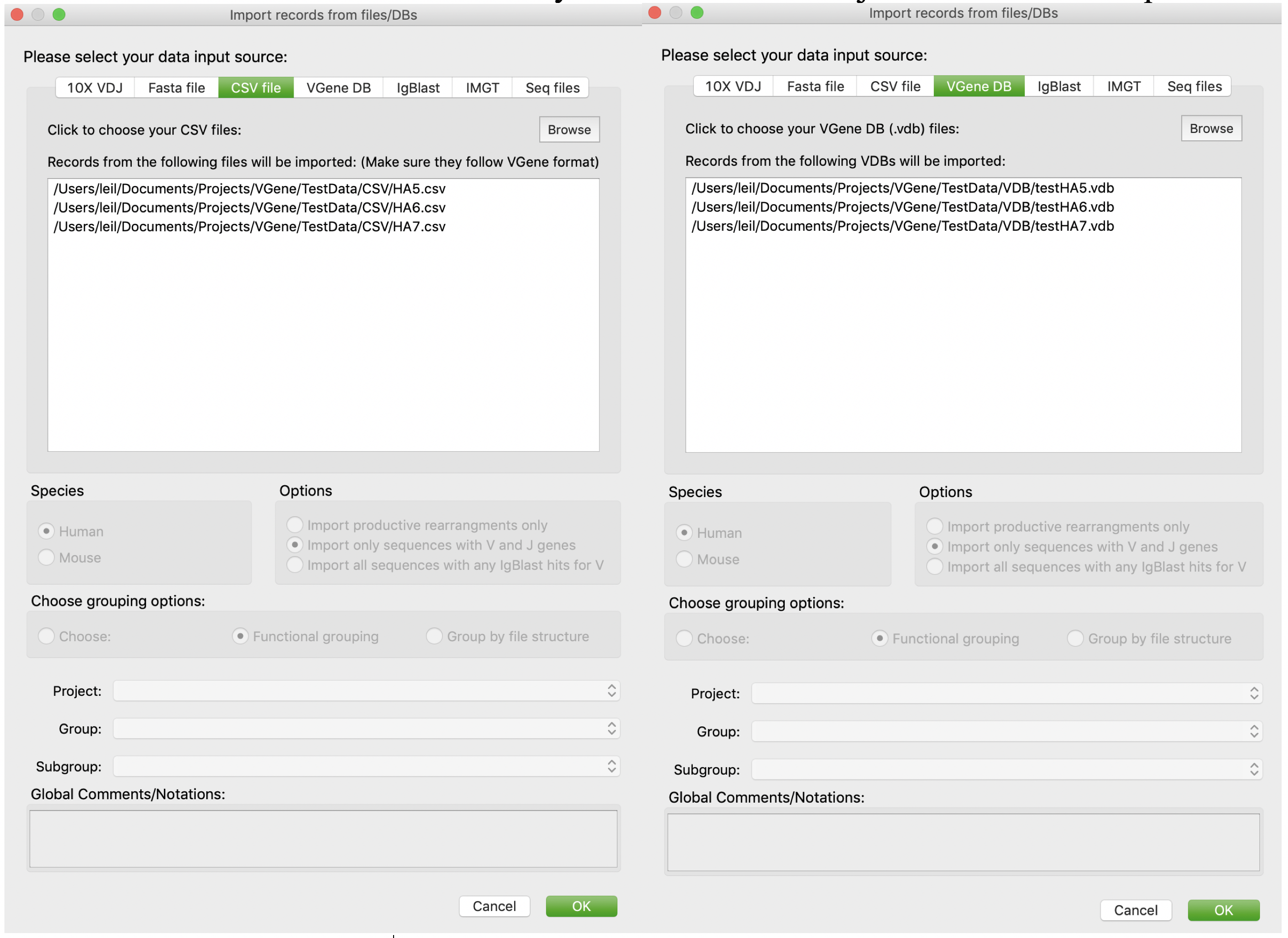
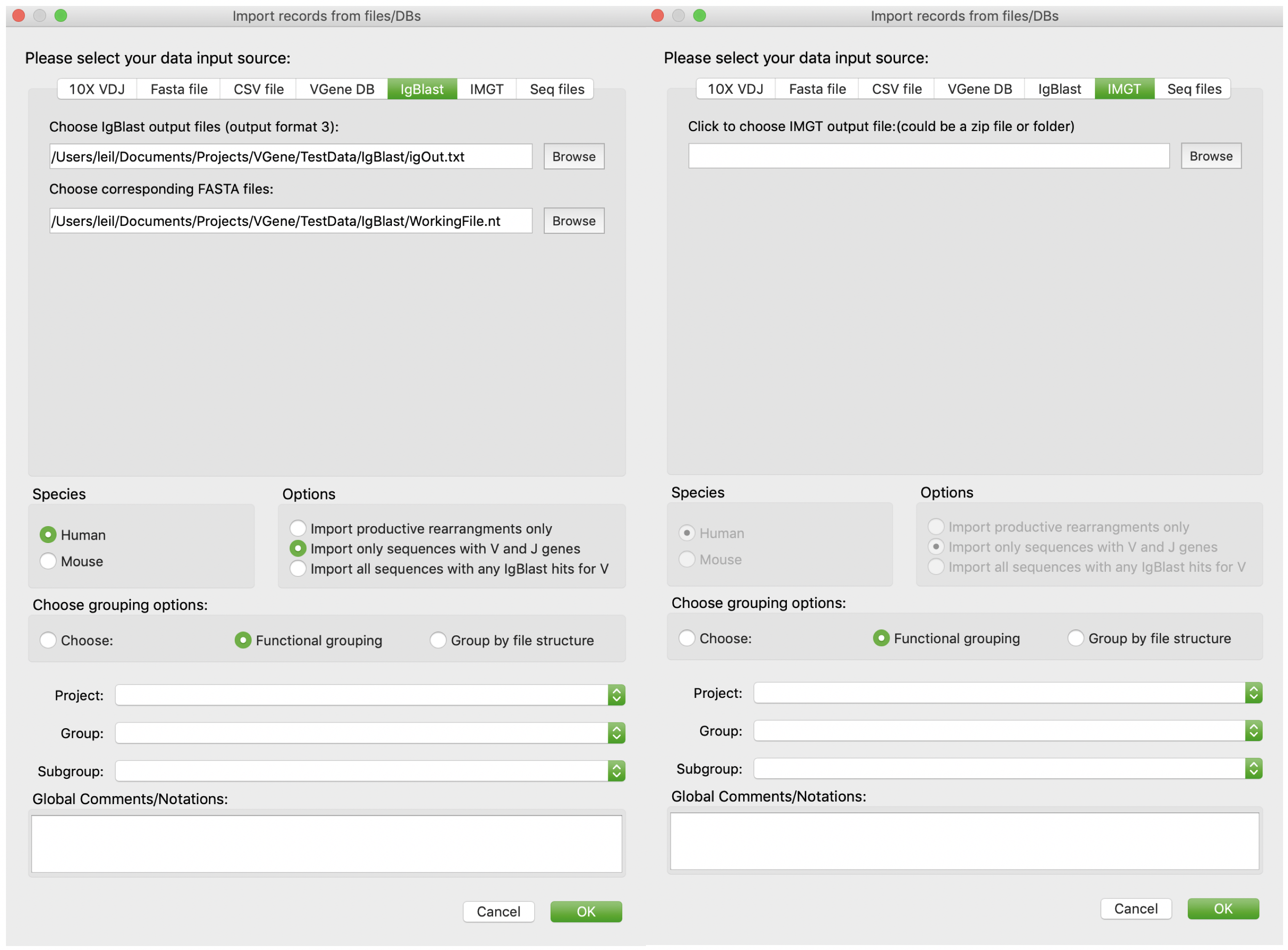
On the other hand, VGenes also allows users to import records from processed results: 1) IgBlast result (outfmt 3) , 2) IMGT results. Last, VGenes also allow users to import records from CSV files exported by VGenes, or VGene database themselves. It enables merging multiple VGene DBs. For example, for a large study with multiple subjects, users can create individual VGenes DB for each subjects, and then merge them into a big database using “import from VDB”.
Import from 10X VDJ
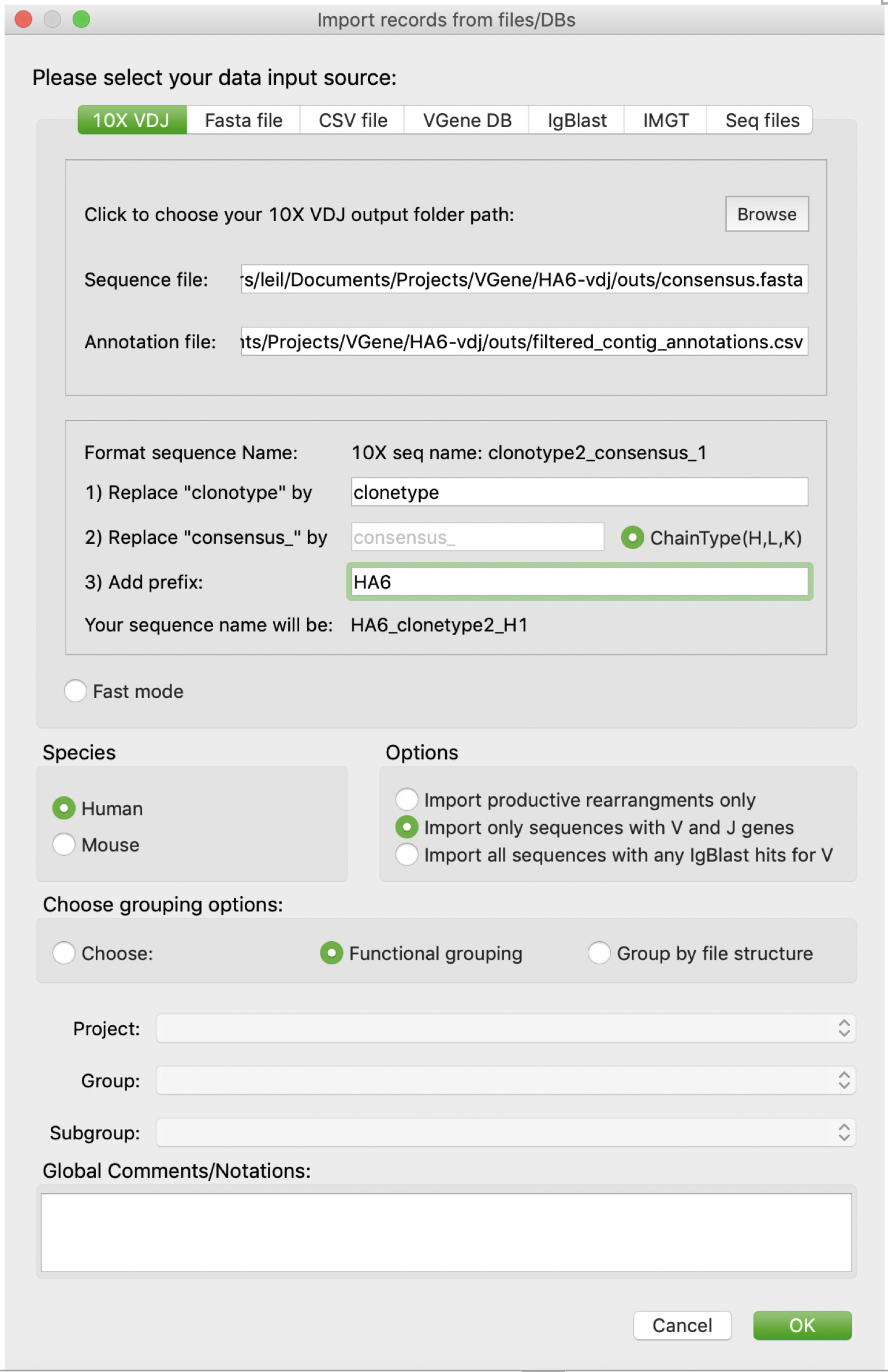
Here, we’d like to demonstrate the data import process using 10X single cell VDJ profiling results (BCR data). First, users should click “Browse” button to browse and determine the path of 10X single cell VDJ profiling results folder. Then VGenes will automatically locate the two required files: consensus.fasta and filtered_contig_annotations.csv. VGenes will pops up an error message if those two files cannot be found under current folder. After that, users are allowed to setup a uniform format for sequence names, e.g. adding prefix that indicating sample origin and/or adding chain type indicators. Last, users are required to choose species, currently only Human and Mouse are supported. After click “OK”, VGenes will start to process the input data. A progress bar will be popped up to indicate the progress.
Import from Fasta files
Users are also allowed to import data from Fasta files. First, users should click “Browse” button to browse and determine the path of Fasta files. Users can either select multiple files at one time, or add multiple files one by one. Users are allowed to remove a file from current candidate list by a double click. The rest settings are as same as those in 10X VDJ page. BTW, file names will be used as prefix to all sequences to indicate sample origins and avoid sequence name redundancy. For example, sequences from file “../HA5.fasta” will have a prefix “HA5_”.
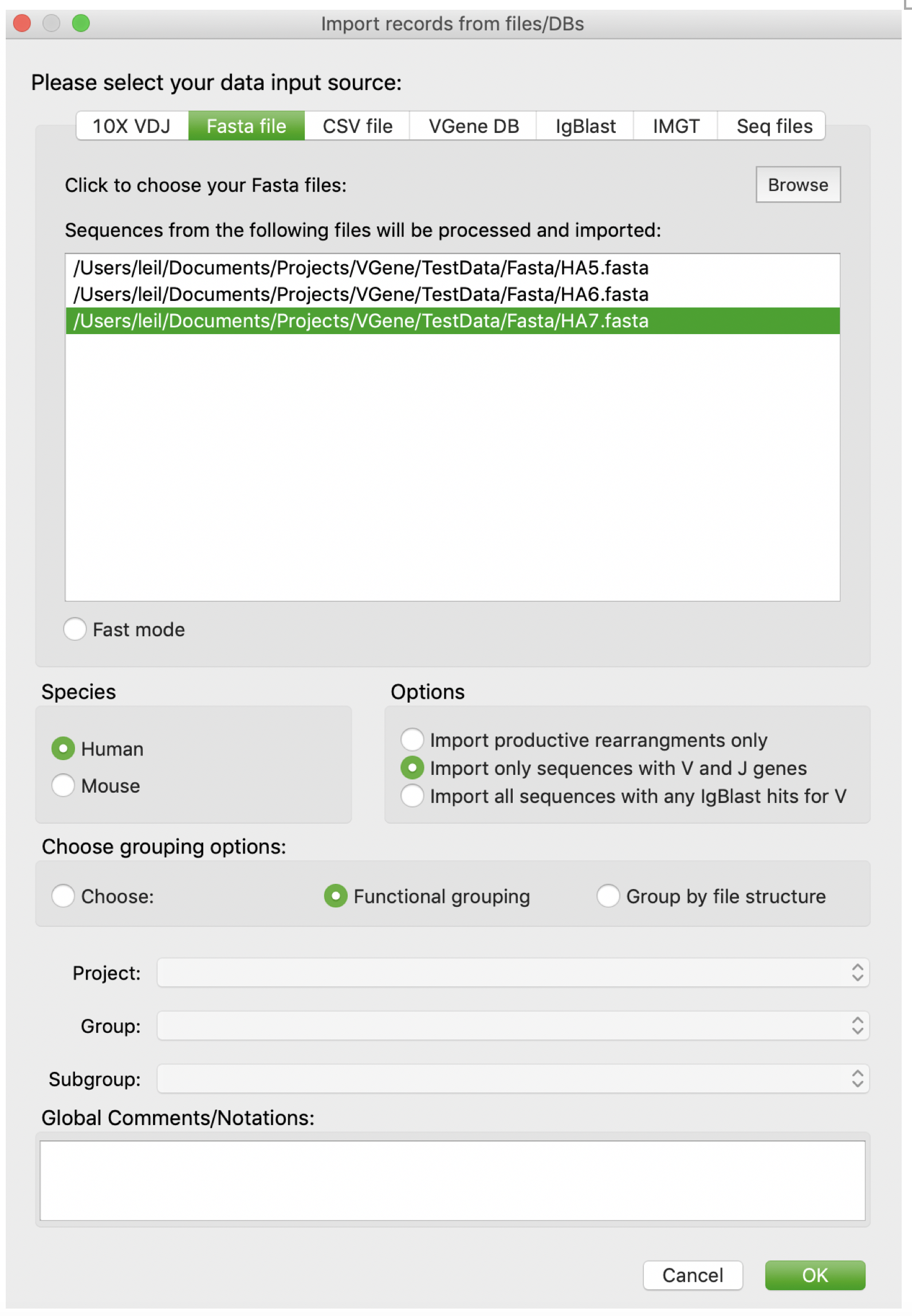
Import from SEQ files
Users are also allowed to import data from SEQ files. First, users should click “Browse” button to browse and determine the path of SEQ file folder. Users can either select multiple folders at one time, or add multiple folders one by one. Users are allowed to remove a file from current candidate list by a double click. The rest settings are as same as those in 10X VDJ page. BTW, folder names will be users as prefix to all sequences to indicate sample origins and avoid redundancy. For example, sequences from folder “../S266Cloning” will have a prefix “S266Cloning _”.
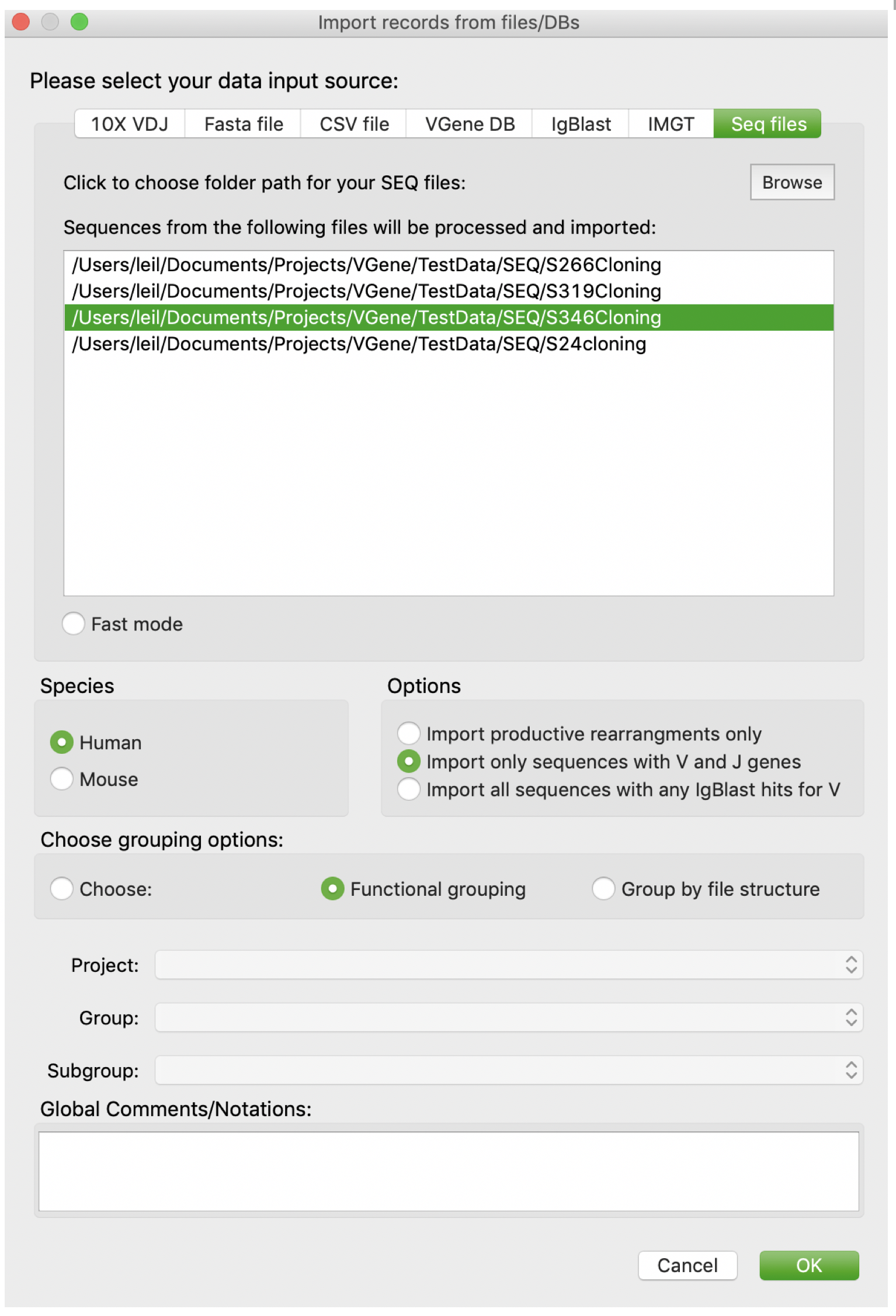
Import from CSV files or VGene DBs
Users are allowed to import records from CSV files or VGene DBs (VDB files). First, users should click “Browse” button to browse and determine the path of CSV files or VDB files. Users can either select multiple files at one time, or add multiple files one by one. Users are allowed to remove a file from current candidate list by a double click. Then just click “OK” to import.
Import from IgBlast or IMGT results
Users are allowed to import records from IgBlast or IMGT results. Users just need to provide the result files then click “OK”.
-
Database annotation- link your VDJ with multi-omics
Users are allowed to annotate their VGenes databases with information from either multimodal profiling (e.g. transcriptome expression, clusters, antigen probe binding) or serological assays (e.g. antigen binding, neutralizing). This function connects V(D)J profiles with genotypic or phenotypic prosperities (especially for single cell data).
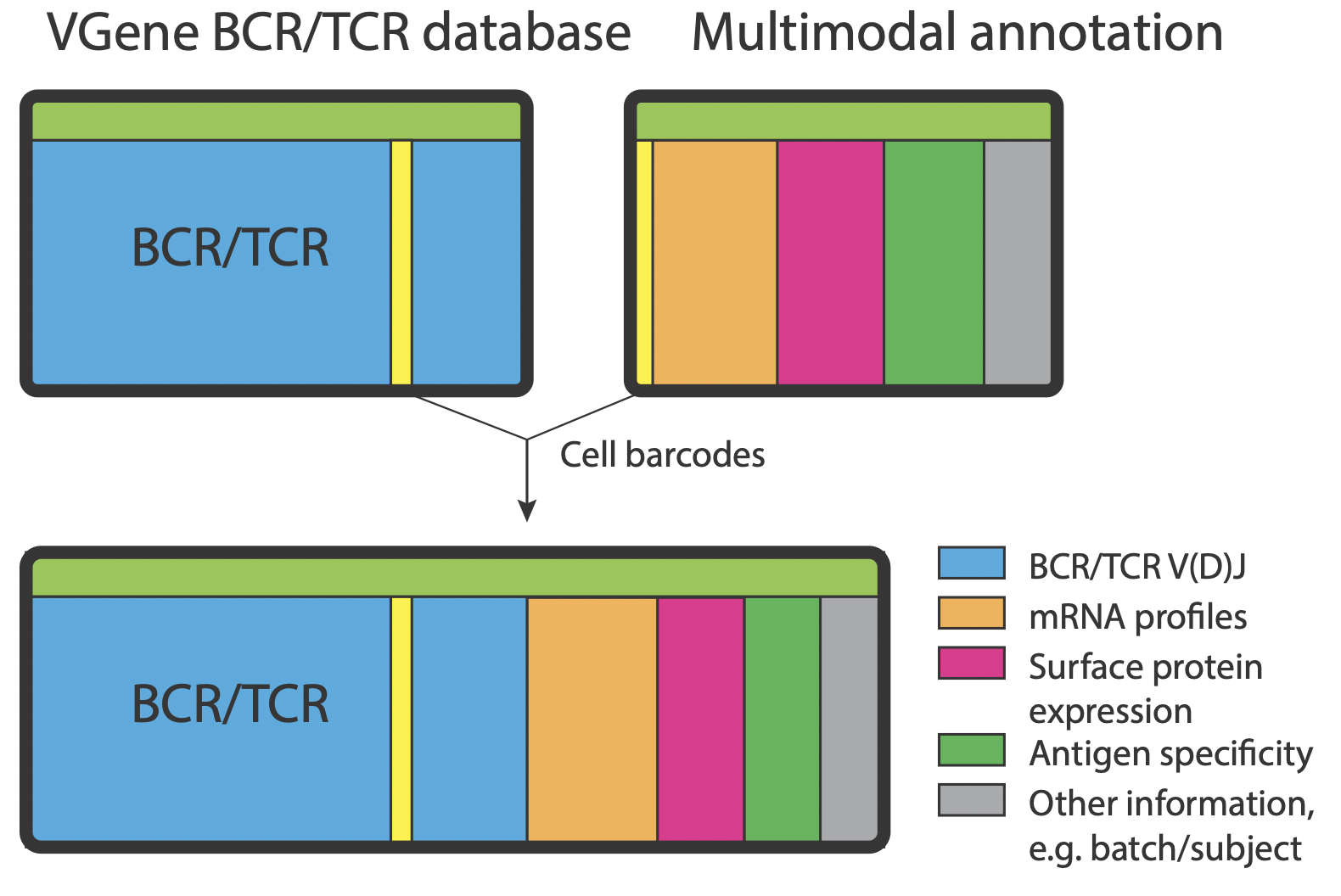
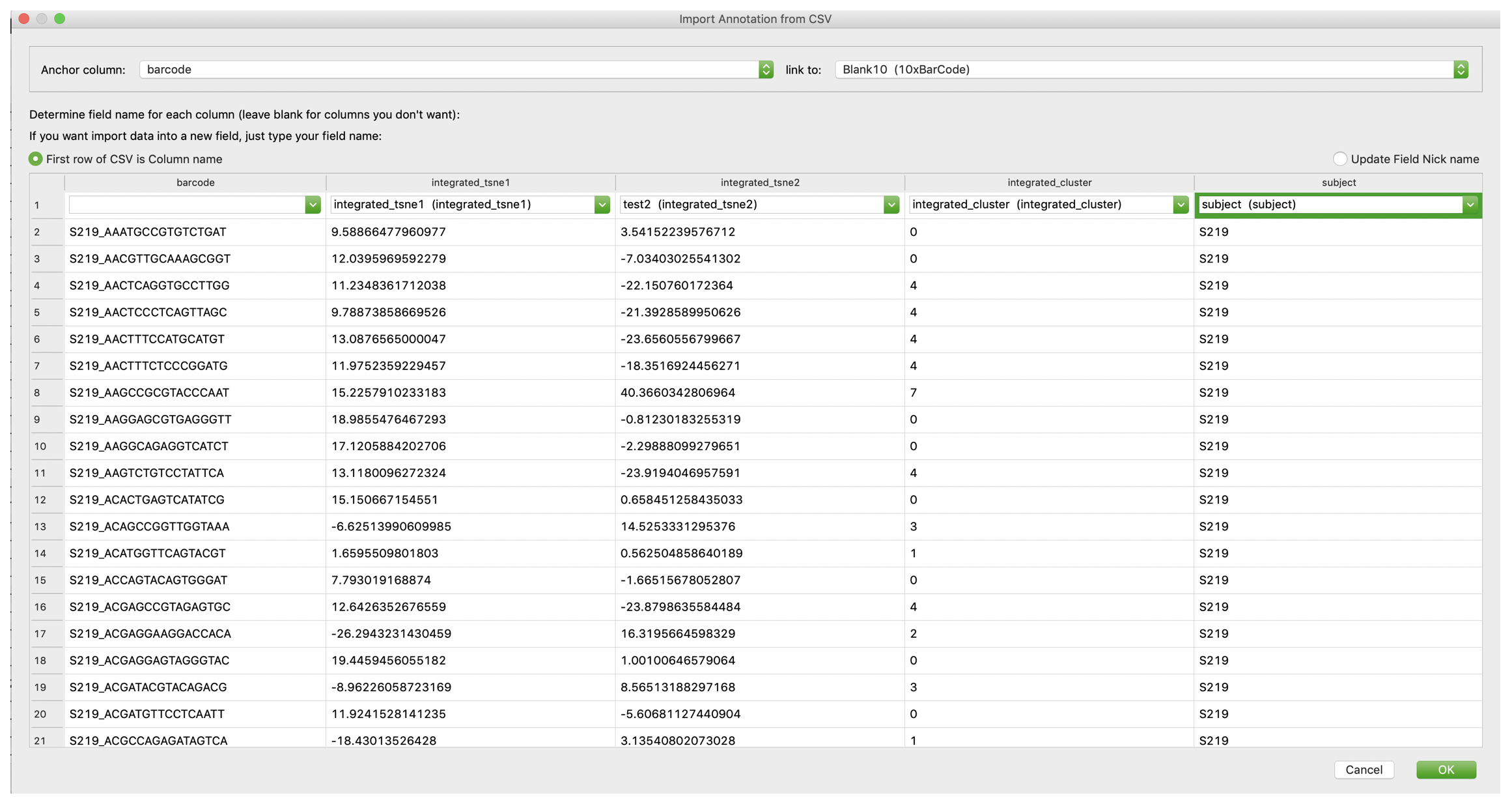
VGenes allows users to import annotation information from CSV files. The CSV file must contain a barcode column that indicate sequence identify. Users could open the annotation dialog by clicking the annotate icon on the toolbar on the main interface, or click Annotate->Import annotate from the menu. Users need to determine the “Anchor column” (barcode column) of the CSV file, and determine the barcode column in the VGenes DB. We pre-defined “Blank10 (10xBarcode)” of VGenes database as the barcode column. VGenes will annotate records by matching information from these columns. Then for all columns need to be imported, users can choose an existing column in VDB to annotate information in that column, or type a new column name to import information to a new column.
BCR/TCR analysis
-
Information display
VGenes integrates all information on a main UI with multiple tabs.
Basic information
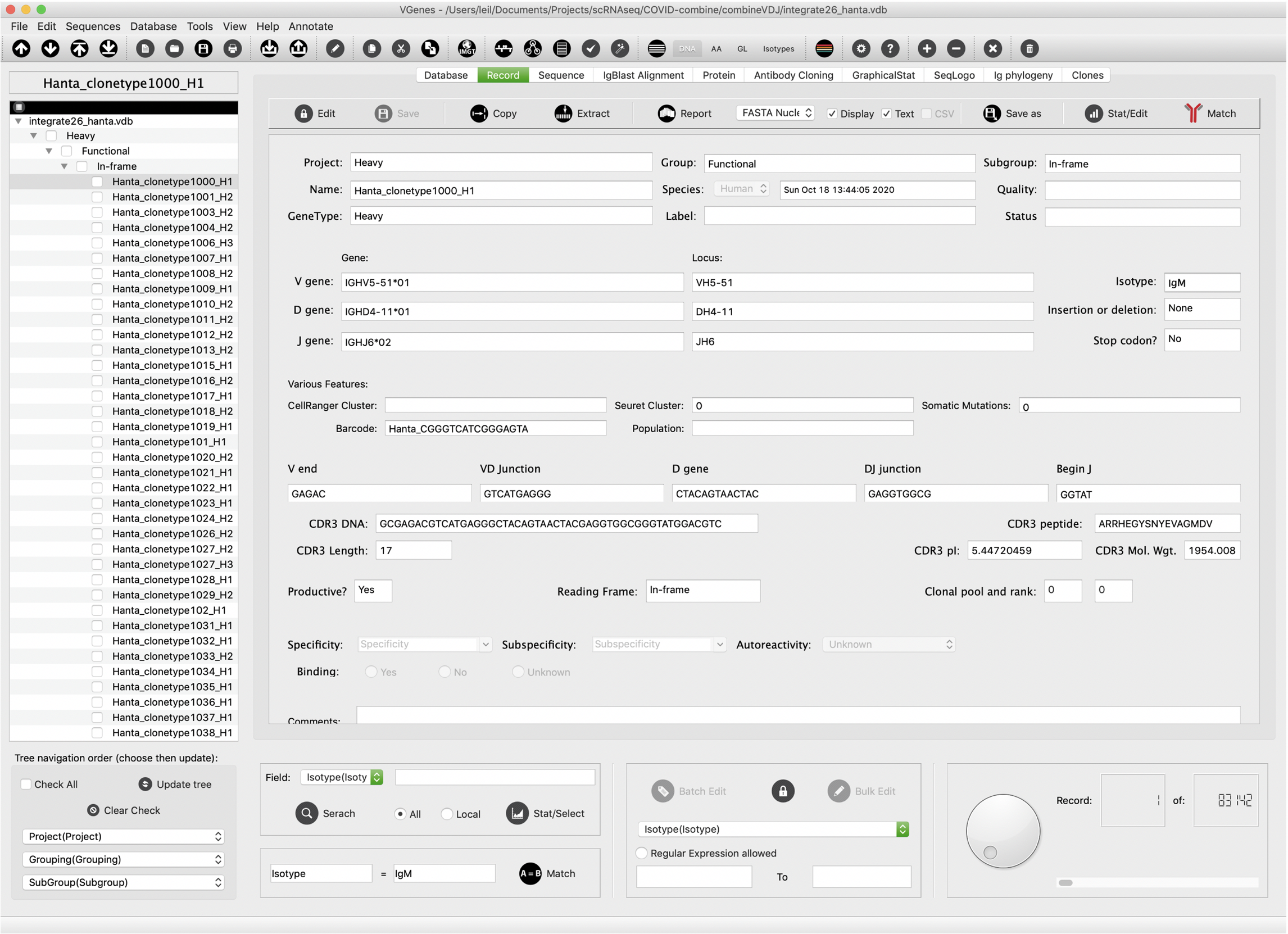
VGenes display basic information of sequences on the “Record” tab. As shown in the following figure, basic information includes: • Sequence name • Gene type • V(D)J genes • Isotype • Barcodes • Transcriptome clusters • CDR3 details • Clone pool information • Other details
Sequence details
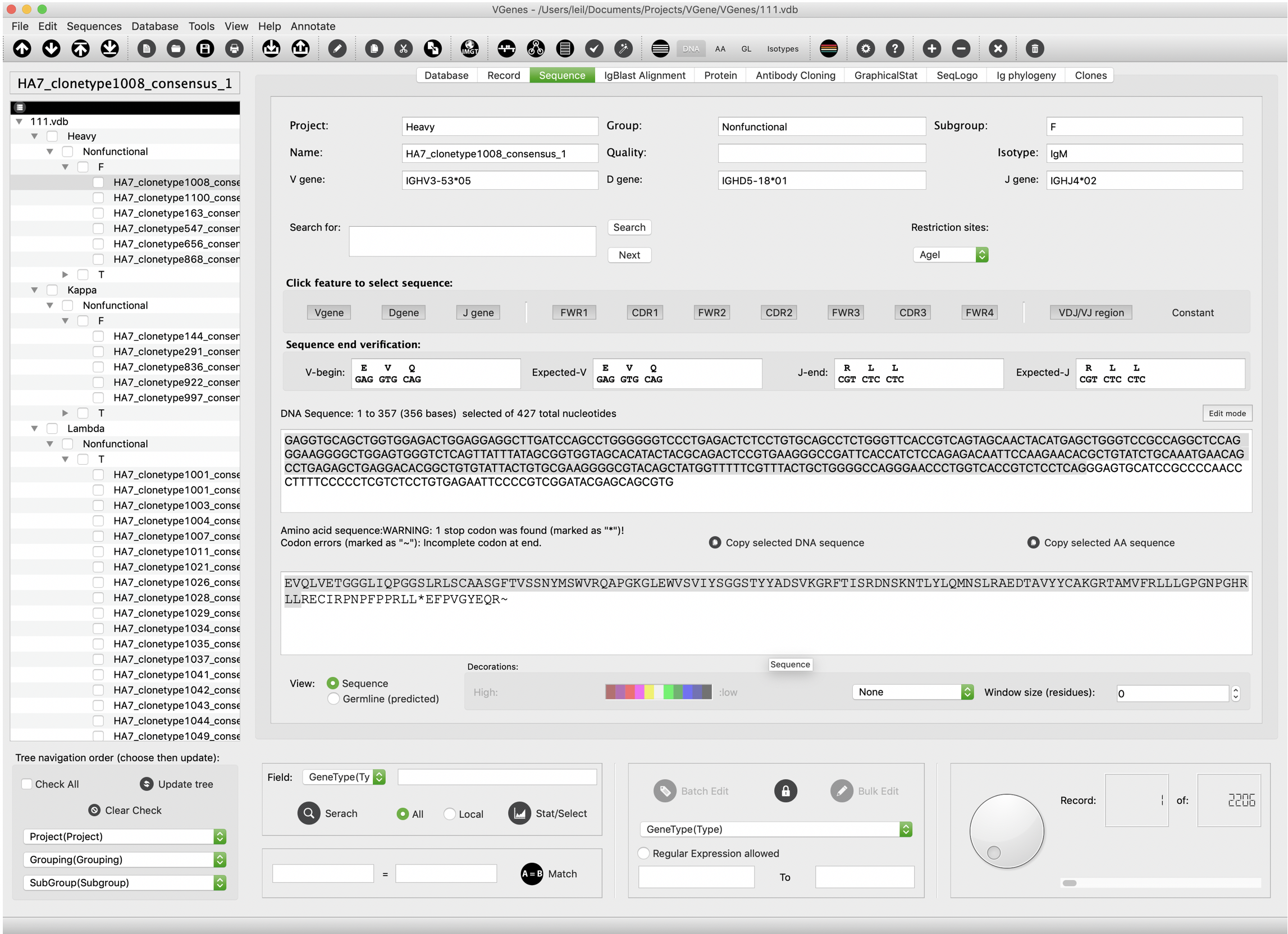
VGenes display sequence details information on the “Sequence” tab. As shown in the following figure, sequence details include: • Sequence name • Gene type • V(D)J genes • Isotype • DNA sequence • Amino acid sequence • DNA germline sequence • Amino acid germline sequence In addition to information display, this tab also allows users to click features (e.g.g V gene, D gene, J gene) to select sequence.
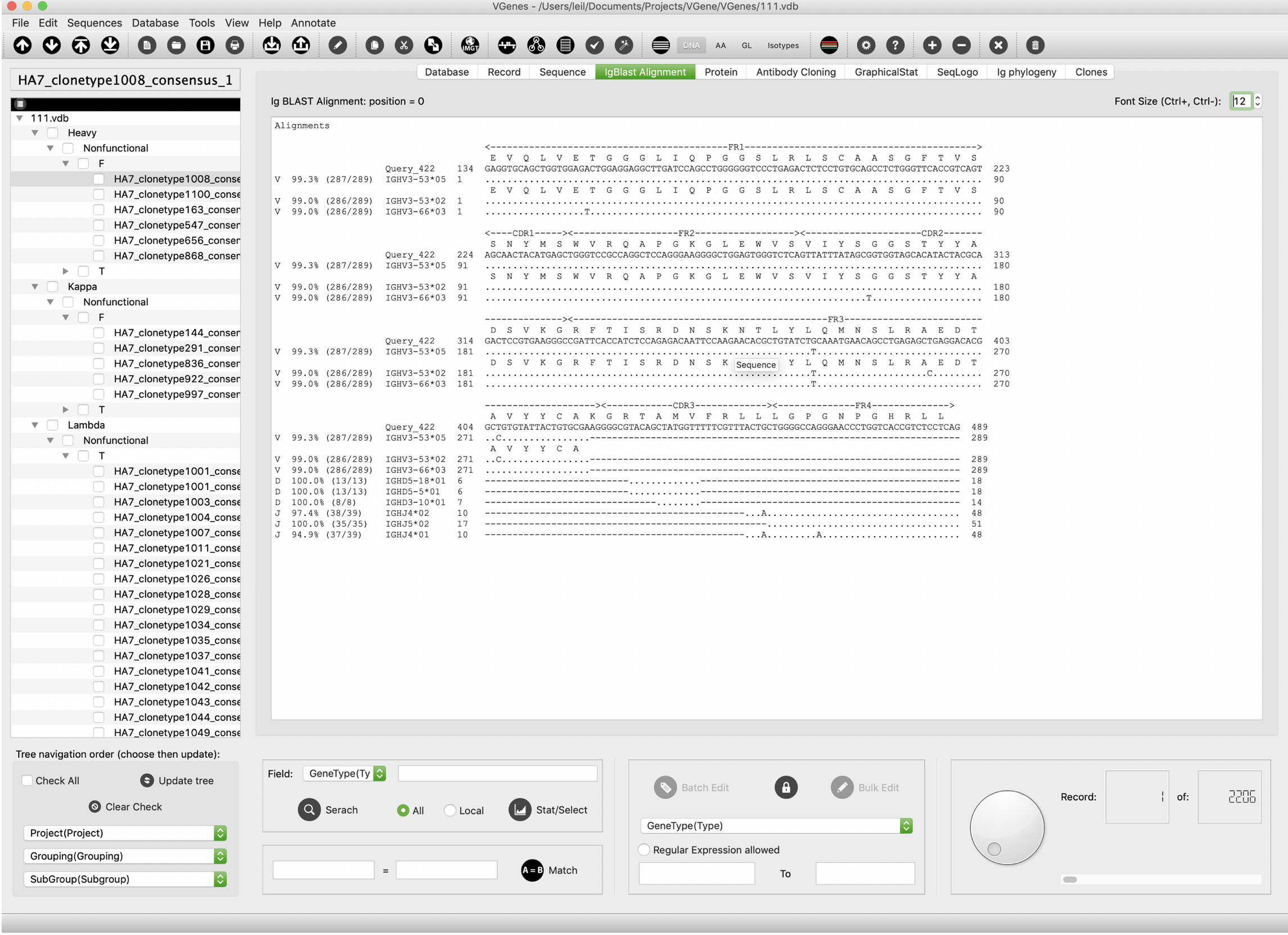
IgBlast alignment
VGenes display IgBlast alignment for current sequence on the “IgBlast Alignment” tab. This page reveals all the details of V(D)J alignment, SHMs and V, D, J locus information.
Records grid view
VGenes displays all the records in a grid viewer on the “Database” tab. Users can browse more detailed information of their Ig genes. Users’ selection on the table viewer will be automatically synchronized to the tree viewer to the left, and vice versa.
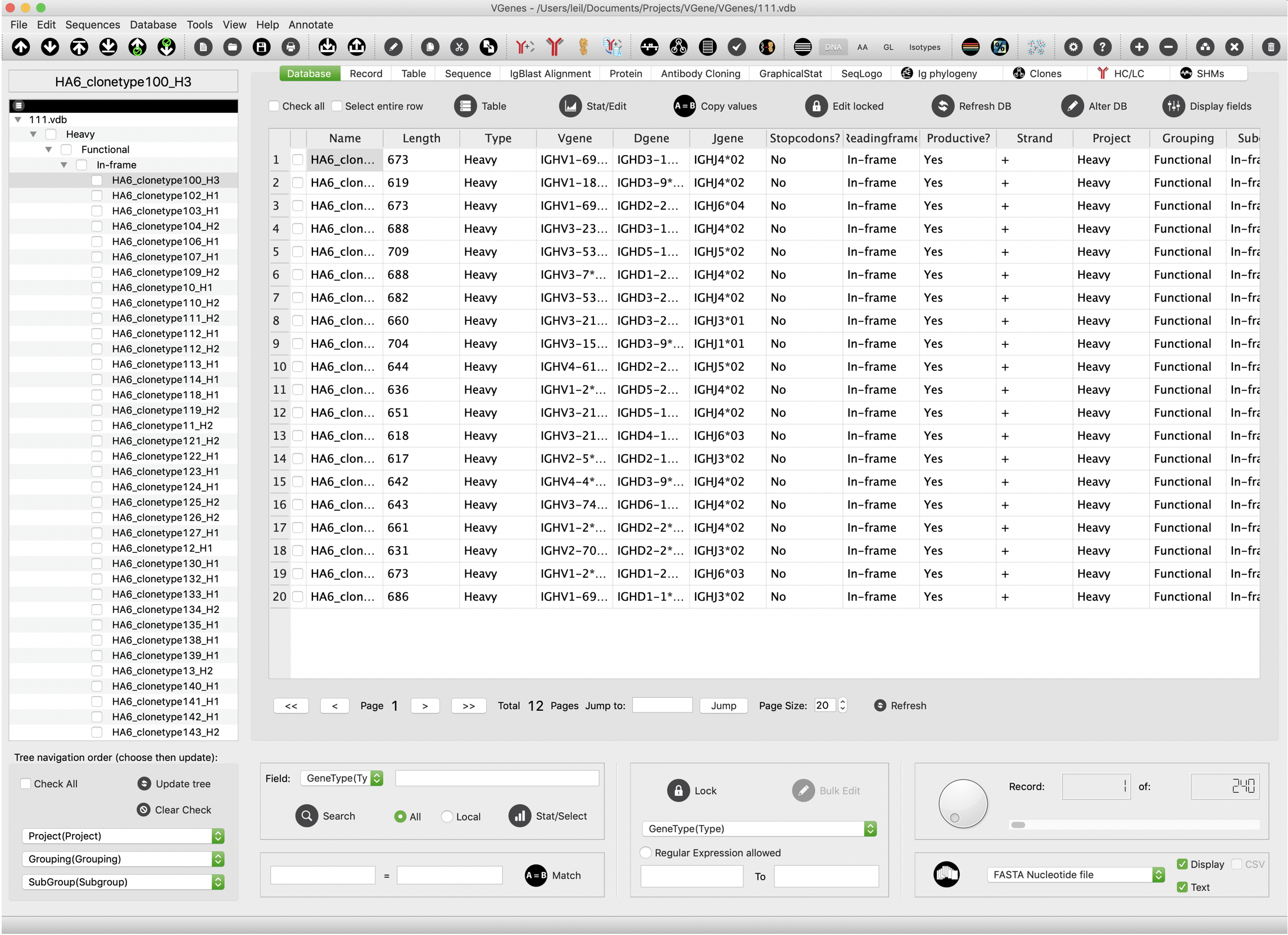
We also integrated lots of functions in this tab page to facilitate data management.

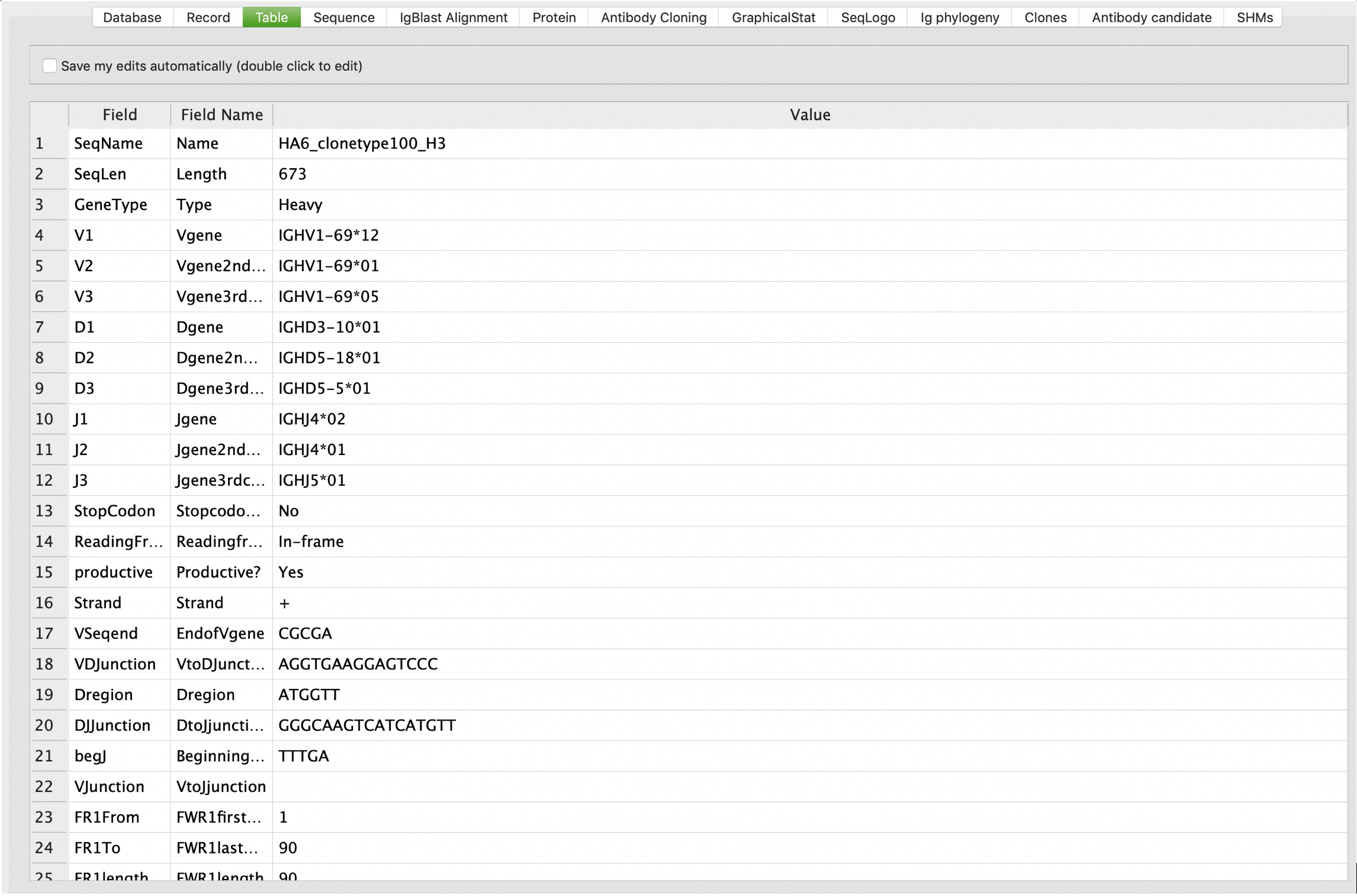
Records table view
VGenes displays all details of current record in a table viewer on the “Table” tab. By checking the “Save my edits automatically” box, VGenes allows users to edit their records.
-
Records edit
UUsers are allowed to edit their records in three ways: edit in table viewer, bulk edit, and advanced edit.
Basic edit
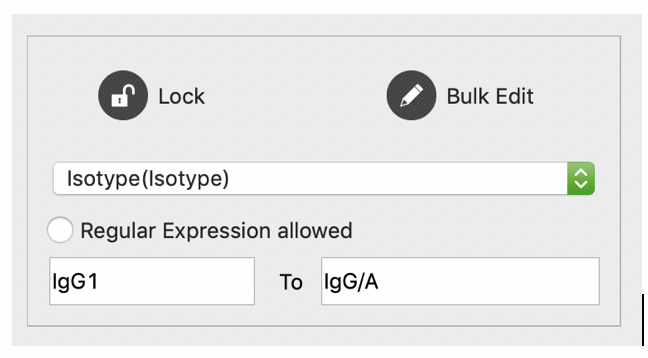
Users can edit records directly in the table viewer (double click to edit, not recommended), or use our bulk edit function. For bulk edit, users should check all records they want to edit first. Then they should set the column name to be edited, the old value and new value. At last click “Bulk Edit” to apply the edit.
Advanced edit
Users are encouraged to edit their records using our advanced edit function. Users can active this function by clicking “Stat/Edit” on the “Record” and “Database” tabs. As shown in the following figures, for each existing value (or value range), users are allowed to update to a new value. Leaving new value blank means unchanged. Here we demonstrate the utility of this function by two examples: for non-numerical factors (e.g. Genetype, Isotype, clusters…), we update all isotypes into two broad categories: IgG/A and IgM/D; for numerical factors (e.g. number of mutations), we update values in different ranges into different levels. This function aims to help users to better and easier annotate their database.
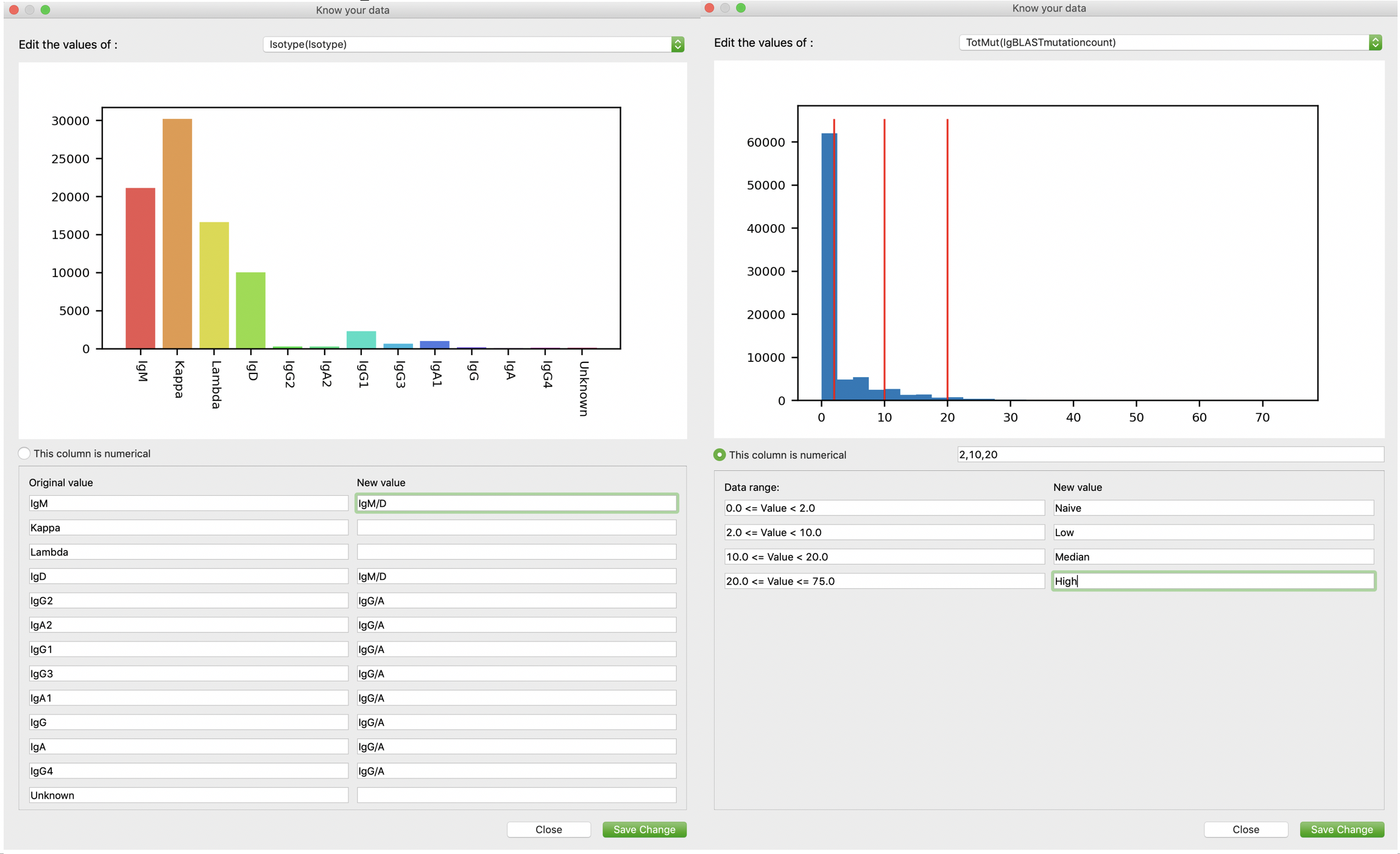
-
Records search/match
It’s important for users to identify all records have a specify property. We designed multiple functions for users to select their interested records.
Basic search
On the left-bottom corner, users can search records with given property. Users can simply select the search field (column), and type the target value, then click “Search” to search. All qualified records will be checked automatically.
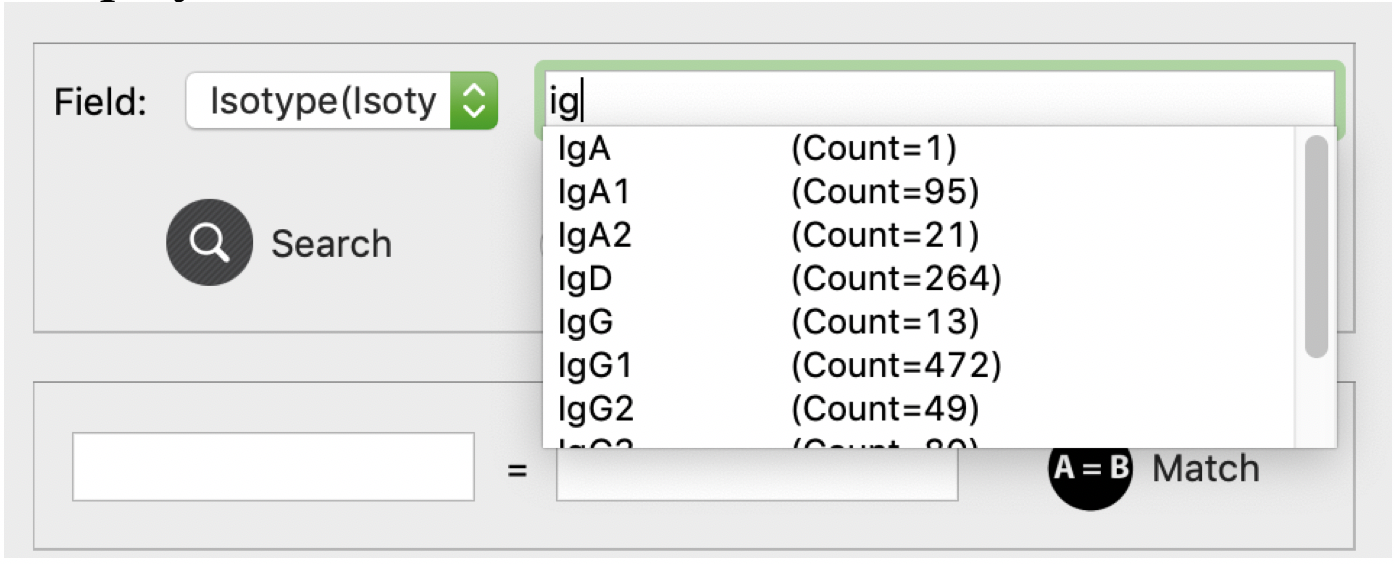
To facilitate users’ operation, we also designed auto-complete functions for the search text field. As shown in below, users are allowed to type part of information and all matched values of current field will be displayed.
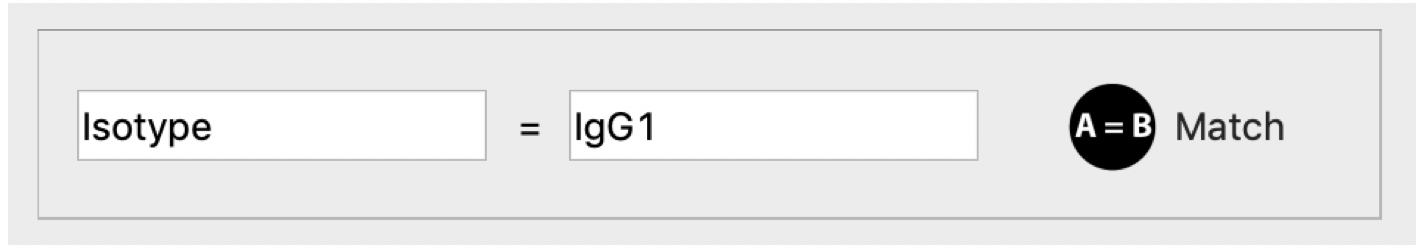
Record match
We developed a function for users to quickly locate all records that share same property with current sequence. Tow inputs for “Match” function will be automatically updated when users click most of fields on the “Record” tab. Then all qualified records will be checked on the tree viewer when users click “Match”.
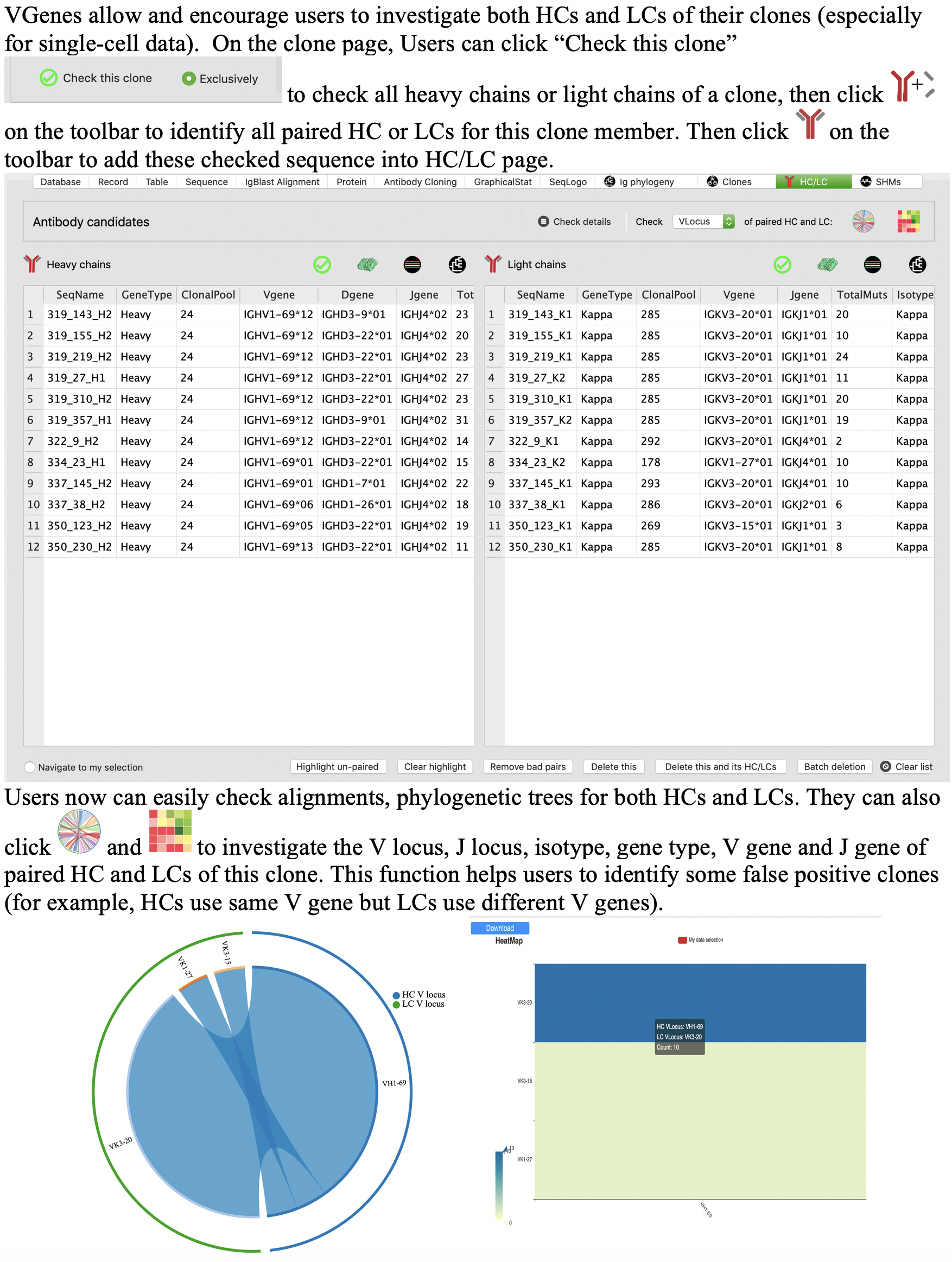
Heavy chain (HC) and light chain (LC) pairing for single-cell data
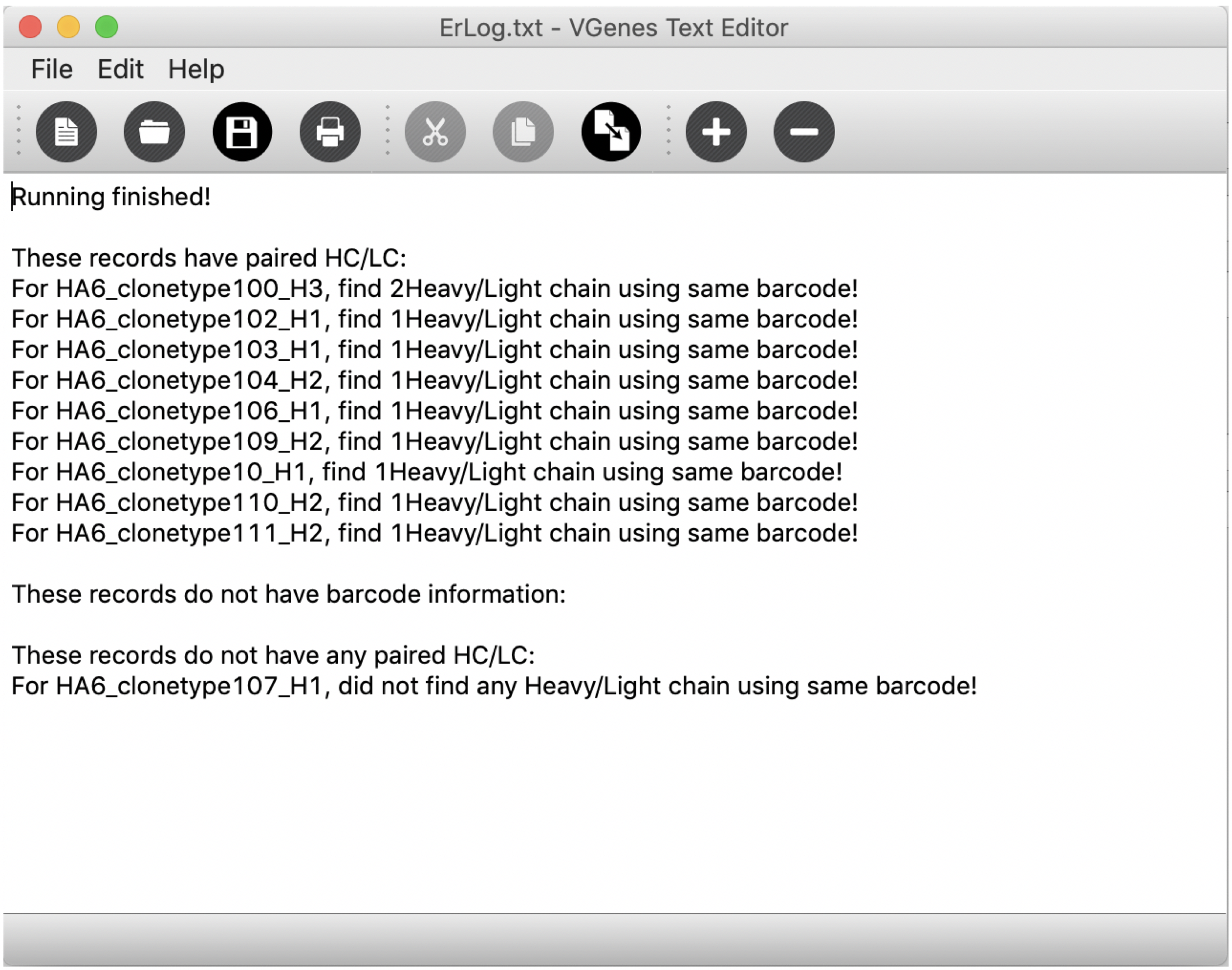
Furthermore, we also provided a function for users to quickly locate all sequences that shares the same barcode with their selected records. This function allows users to quickly find the paired heavy/light chains. Users can click the pairing icon in the toolbar to match paired heavy/light chains for current checked records. If there is not records have been checked, this function will match paired records for current mouse pick (current record displayed in “record” tab). This function will check all qualified records and return a detailed list with how many paired records have been matched for each query record (as shown in the following figure).
VGenes also provides a function for users to identify paired HC/LCs and export them into a new database. Users can click the following icon in the toolbar to active this function.
Advanced search/match
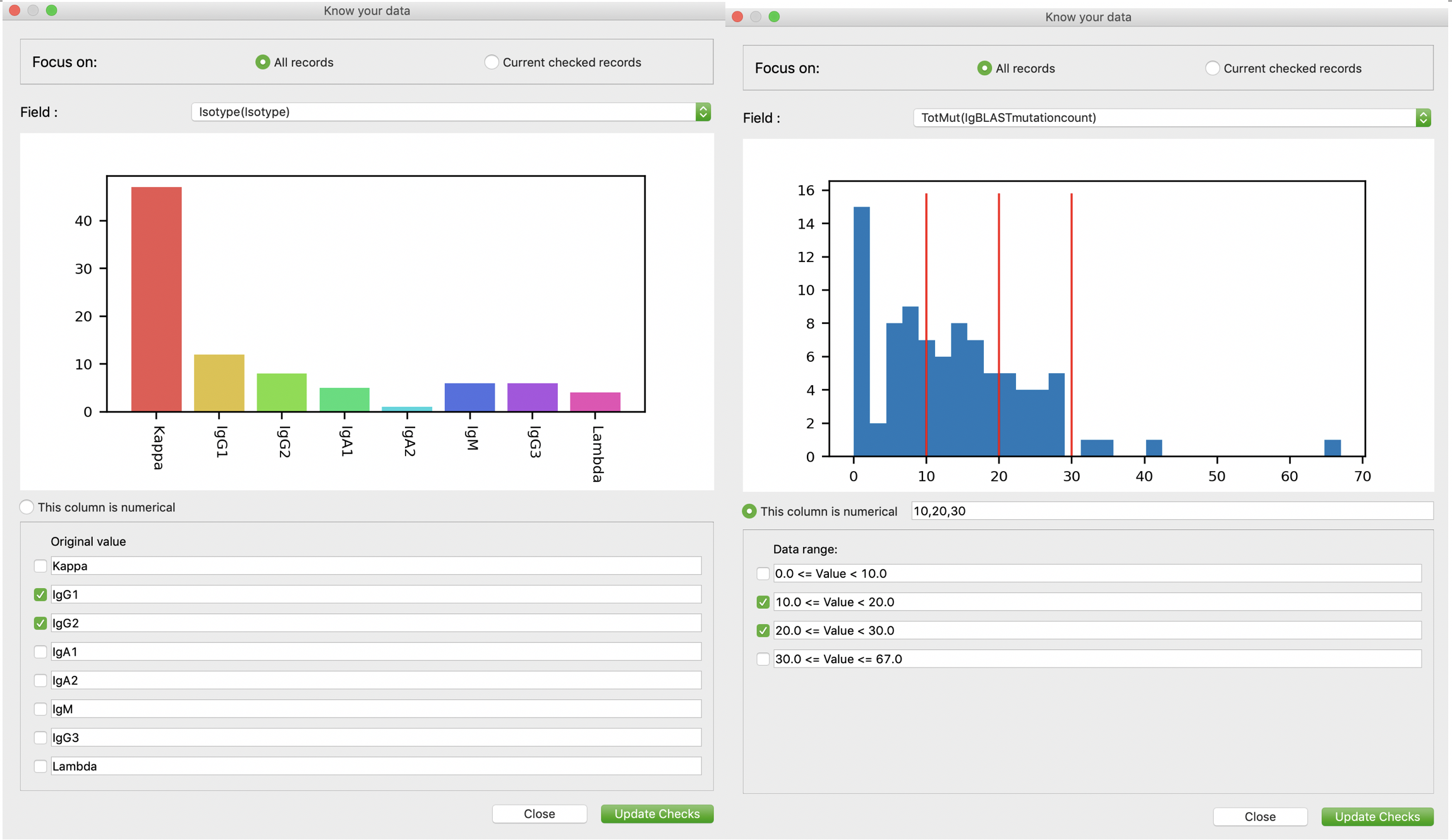
Like we did for “advanced edit”, we developed advanced search/match function in VGenes too. Users are encouraged to search their records using our advanced search/match function. Users can active this function by clicking “Stat/Select” on the left-bottom corner of main UI. As shown in the following figures, users can check the checkboxes for values they want (left panel). Furthermore, for numerical field, for example number of mutations, users can click “this column is numerical” to display the distribution on a histogram, determine cutoffs to divide data into multiple ranges, and then check the ranges they want (right panel). Then users can click “Update checks” to update checks. As users may noticed, there are two different focuses on the top: “All records” and “Current checked records”. Users can focus on current check records to select records from current checks. This enables users to select their interested records using multiple filters.
Update search on graphical statistic
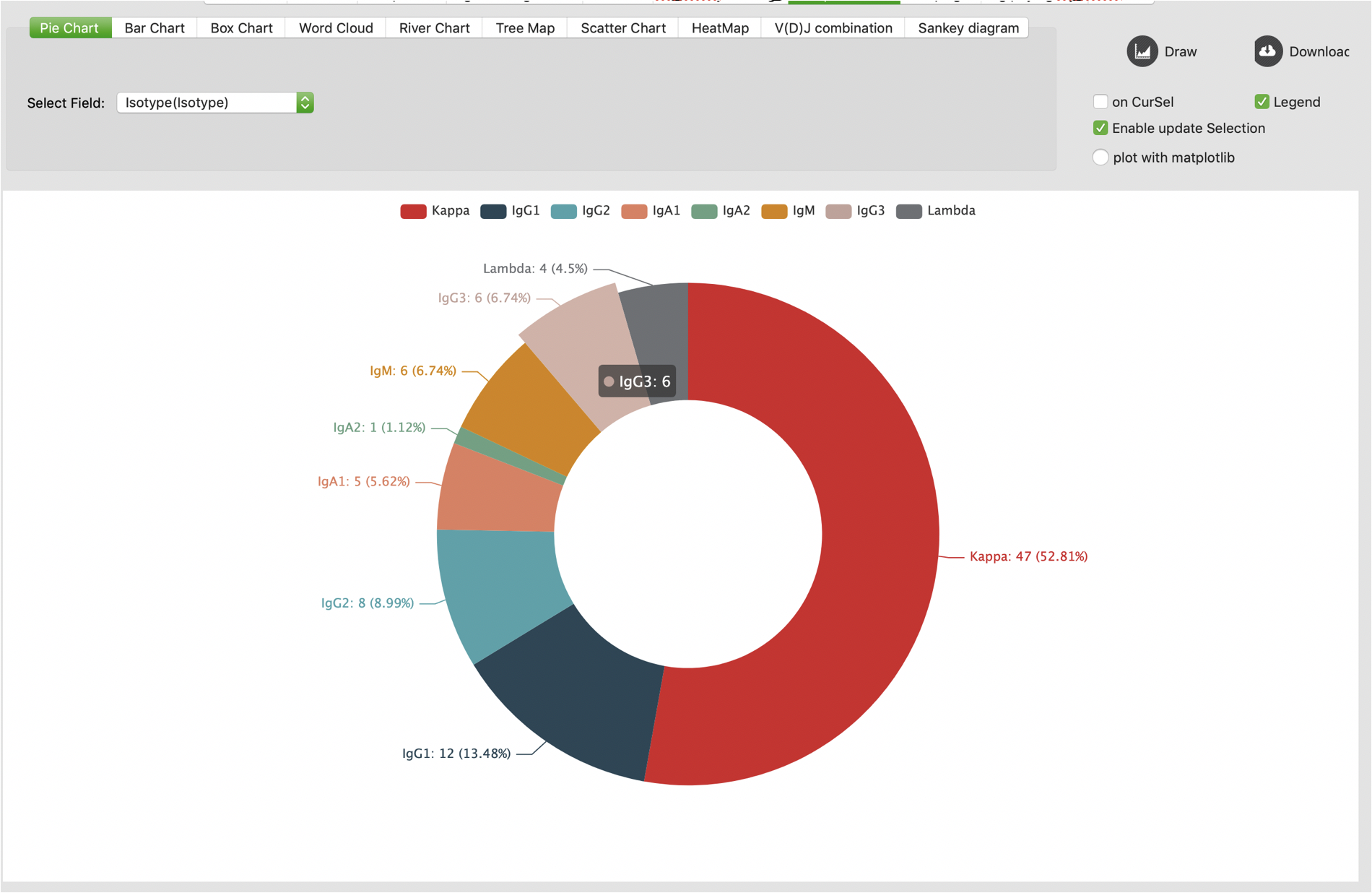
Users are also allowed to update selections on graphical statistic (pie chart, bar chart, box chart, scatter chart, VDJ combination heatmap). To enable this function, users should check the “Enable update Selection” box on “GraphicalStat” tab. Notably, this function is based on current selection by default, it will clear all checks if there is no qualified record from current checks. For example, users can click “IgM” on a pie chart to check all IgM records. Then users click “IgD” on the pie chart. Because there is no qualified record in current selections, the checked records will be reset. Then users should click “IgD” again to select all IgD records.
-
Graphical statistic
We developed variety of functions for graphical statistic. These functions allow users to better understand and investigate their data.

Pie chart
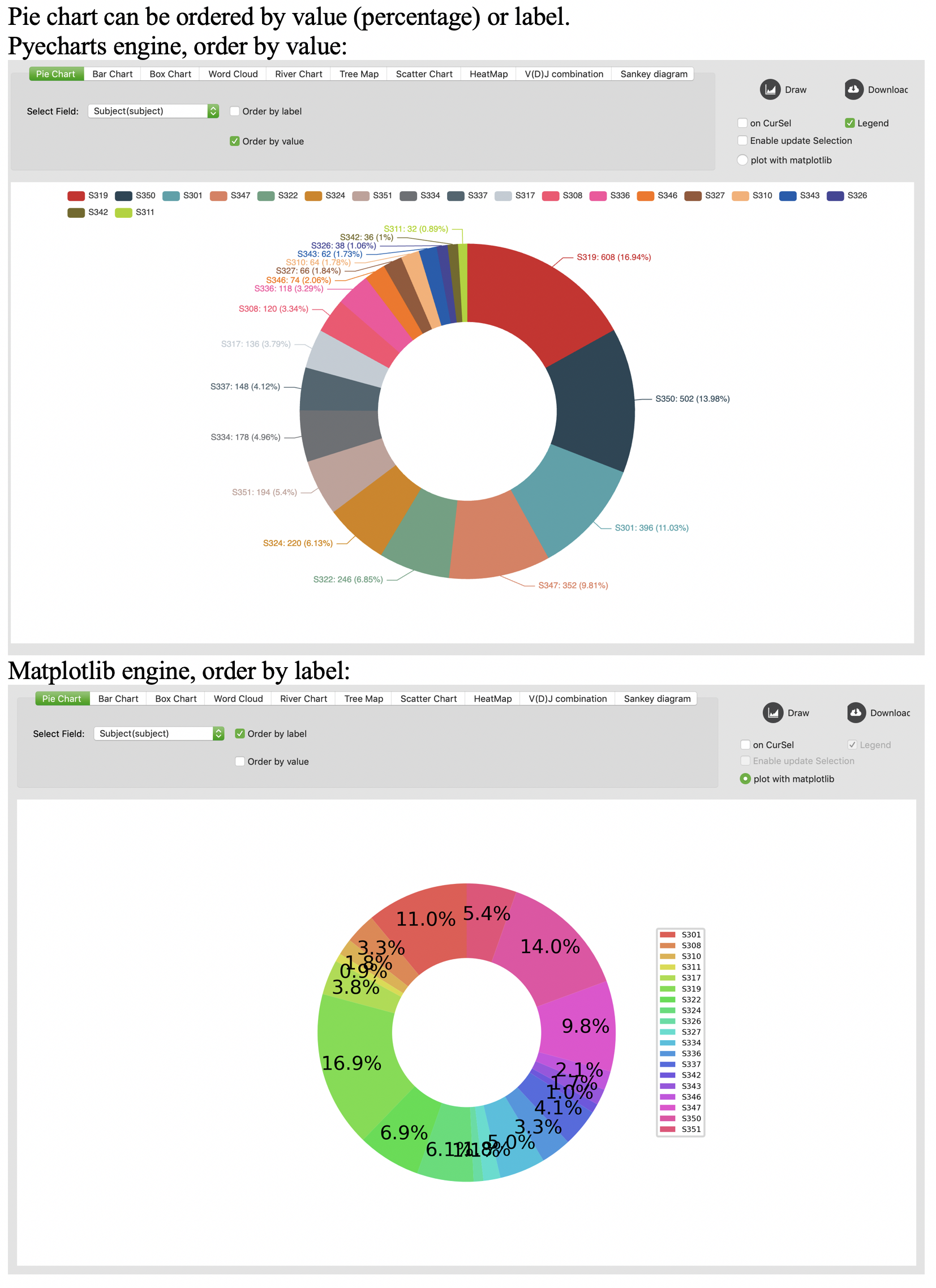
Bar chart
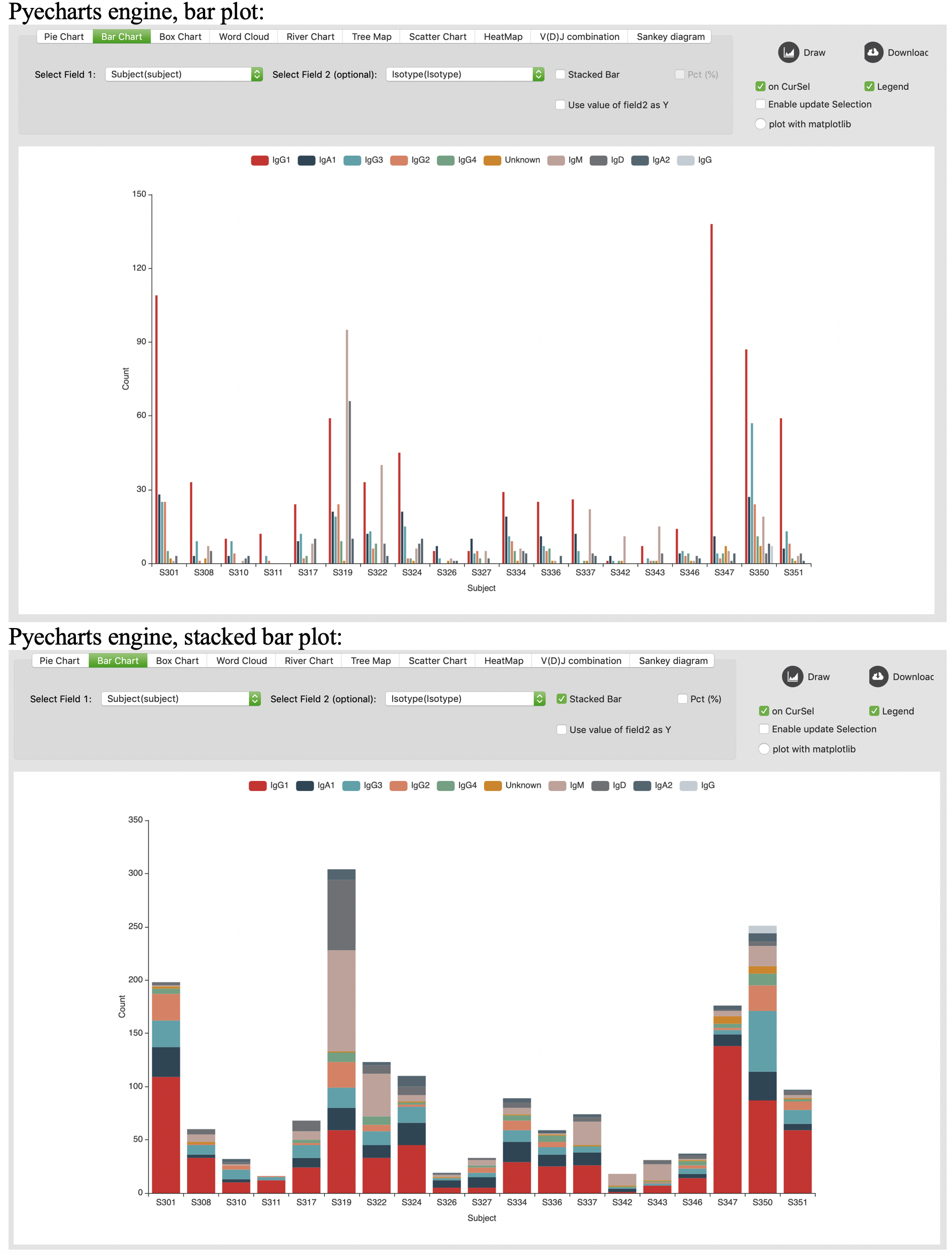
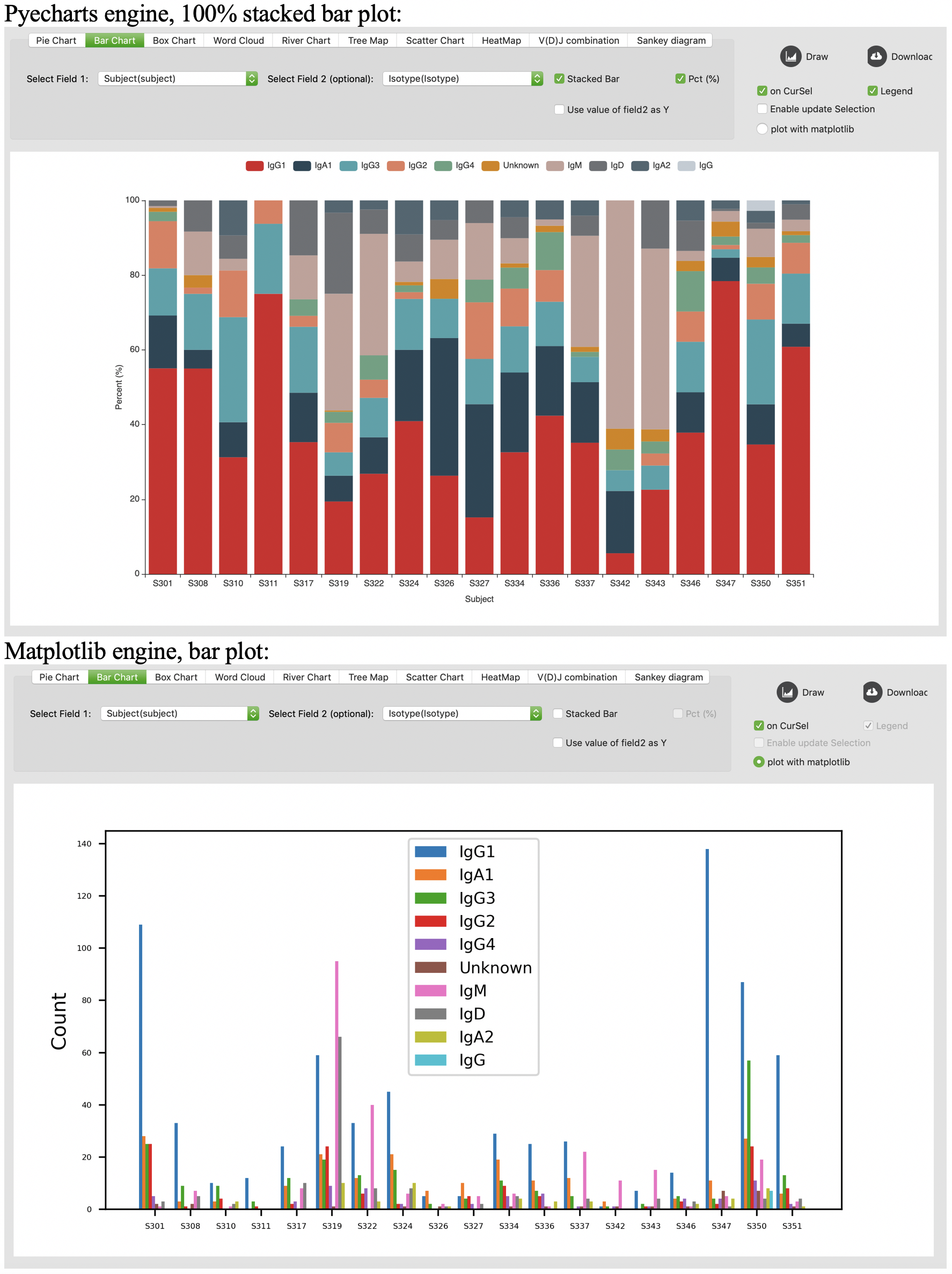
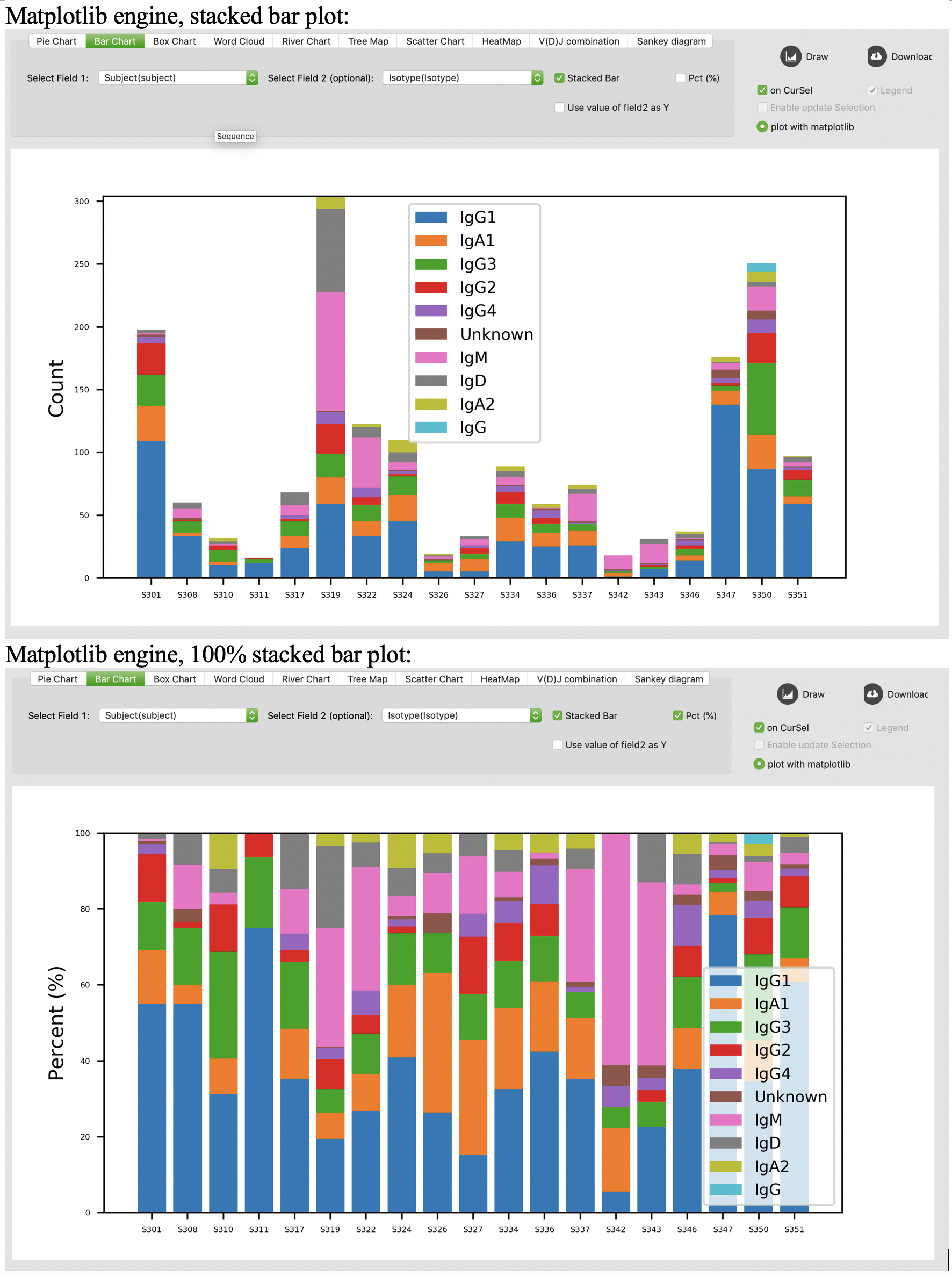
Box chart
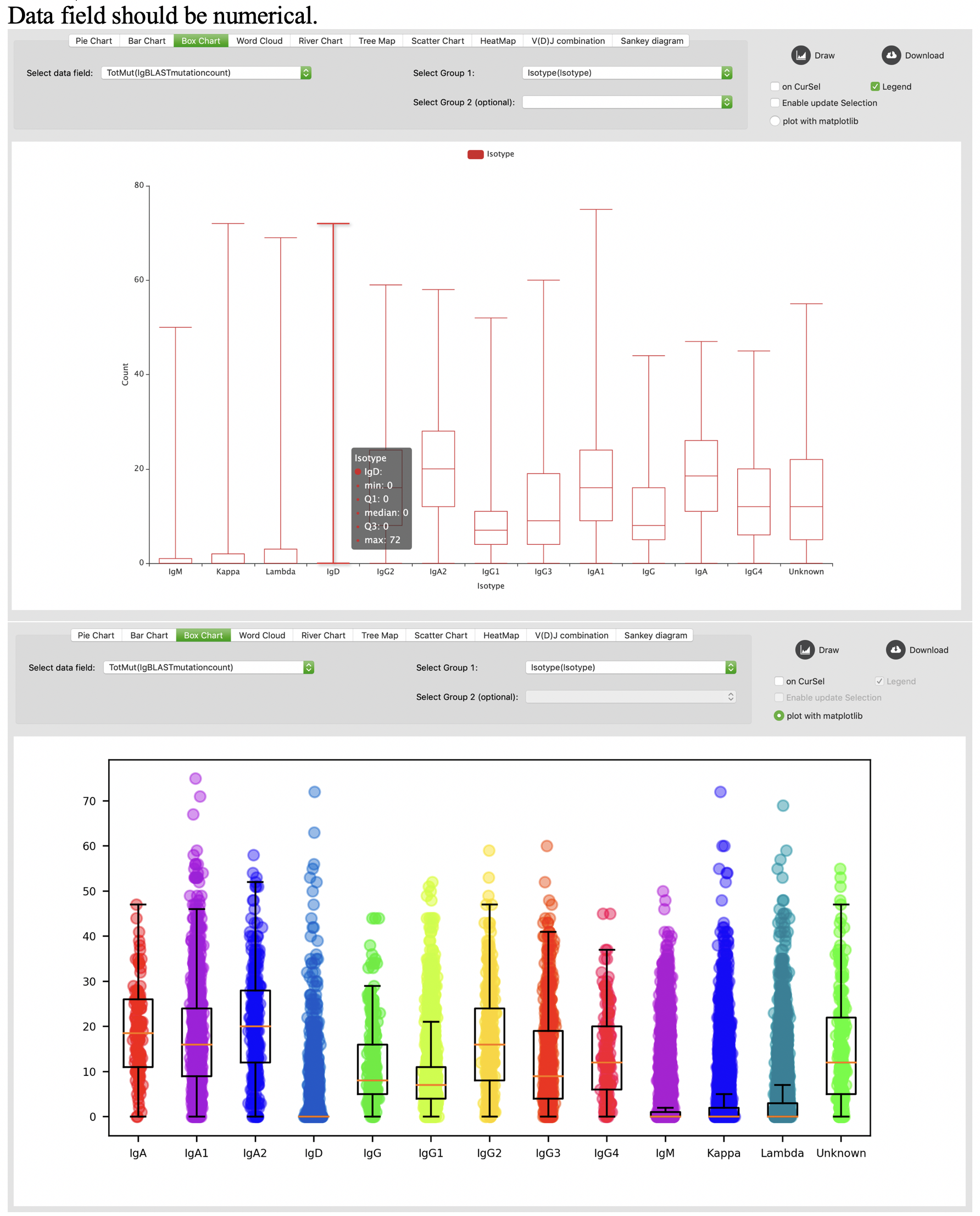
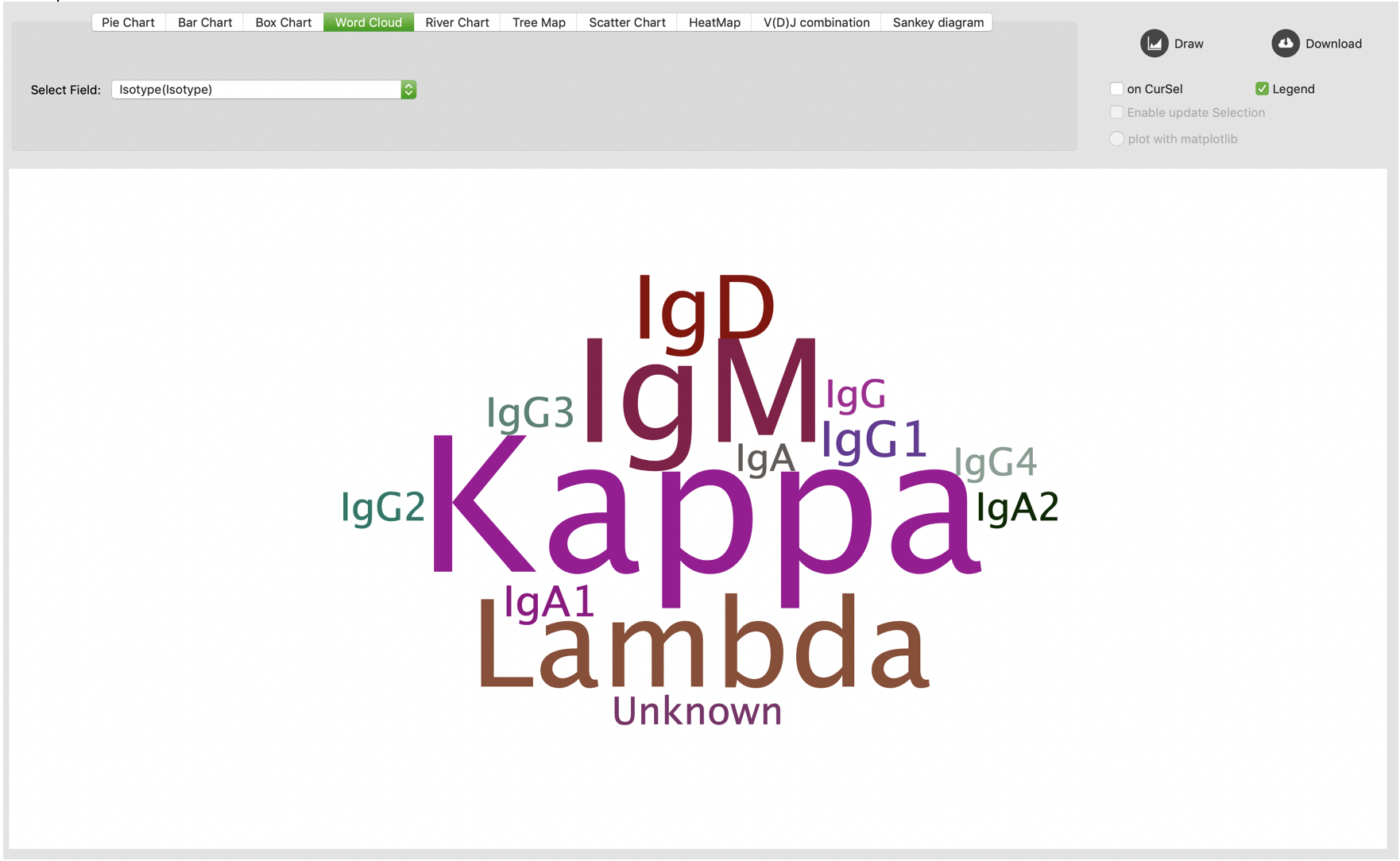
Word cloud
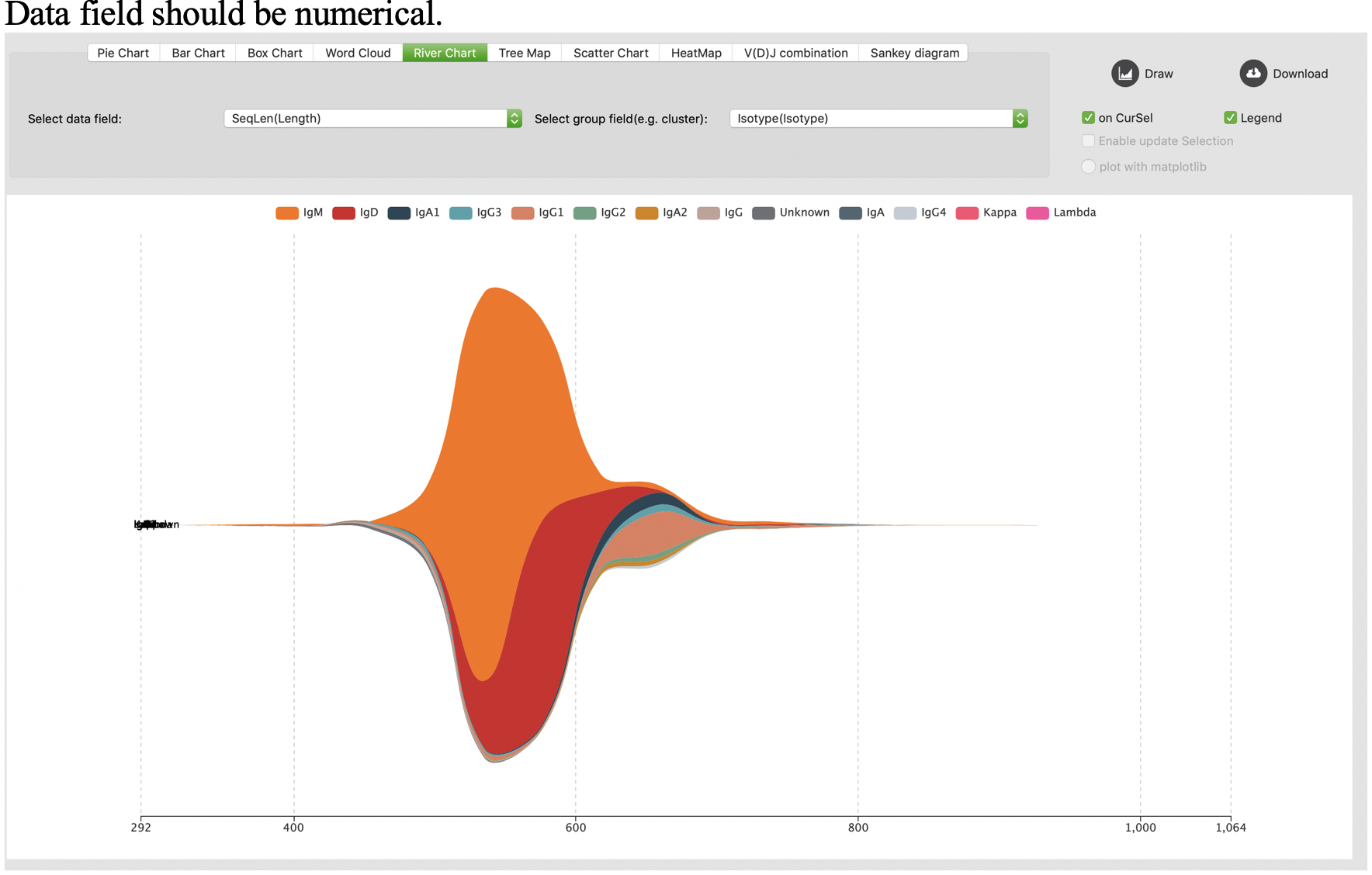
River chart
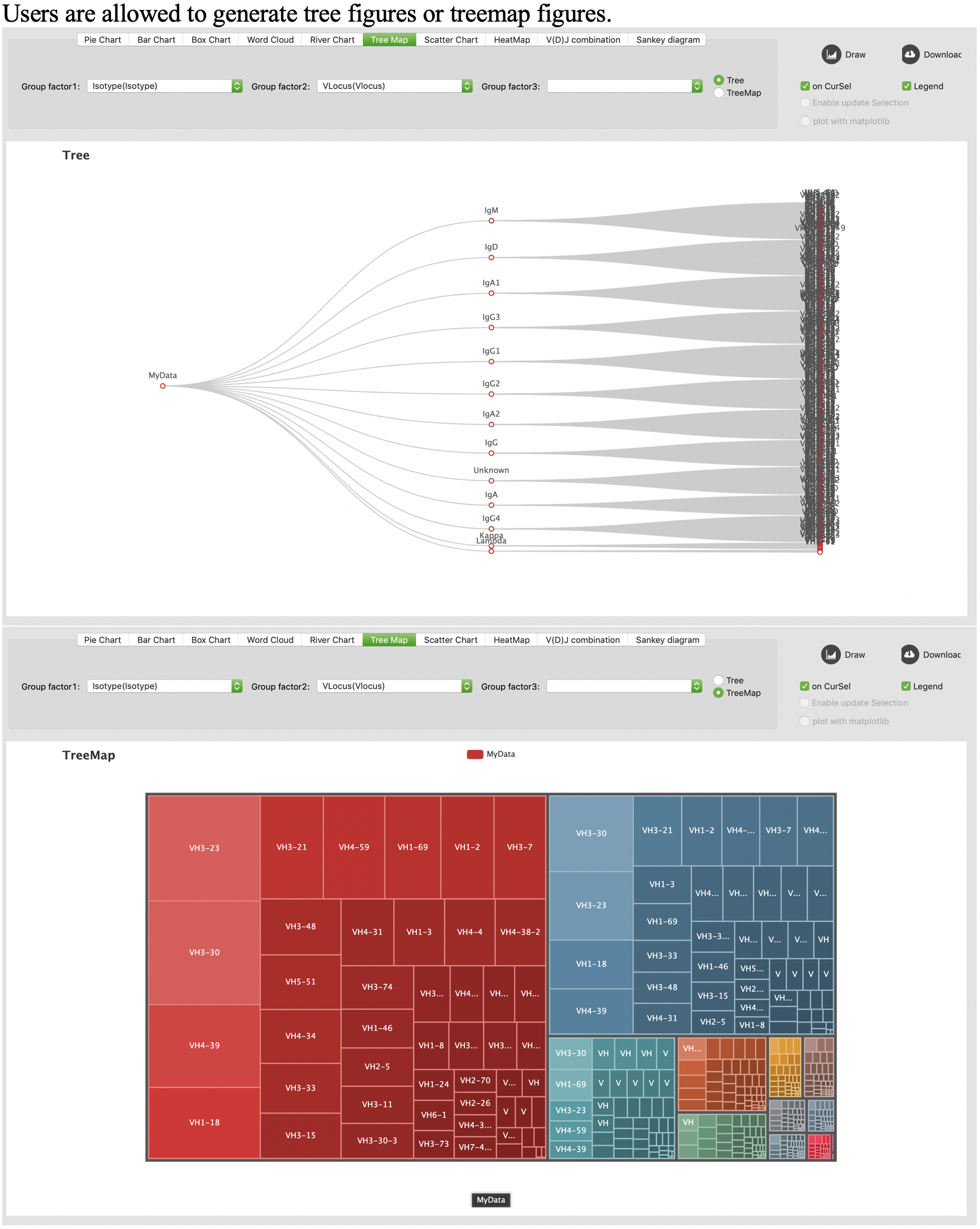
Tree map
Scatter chart
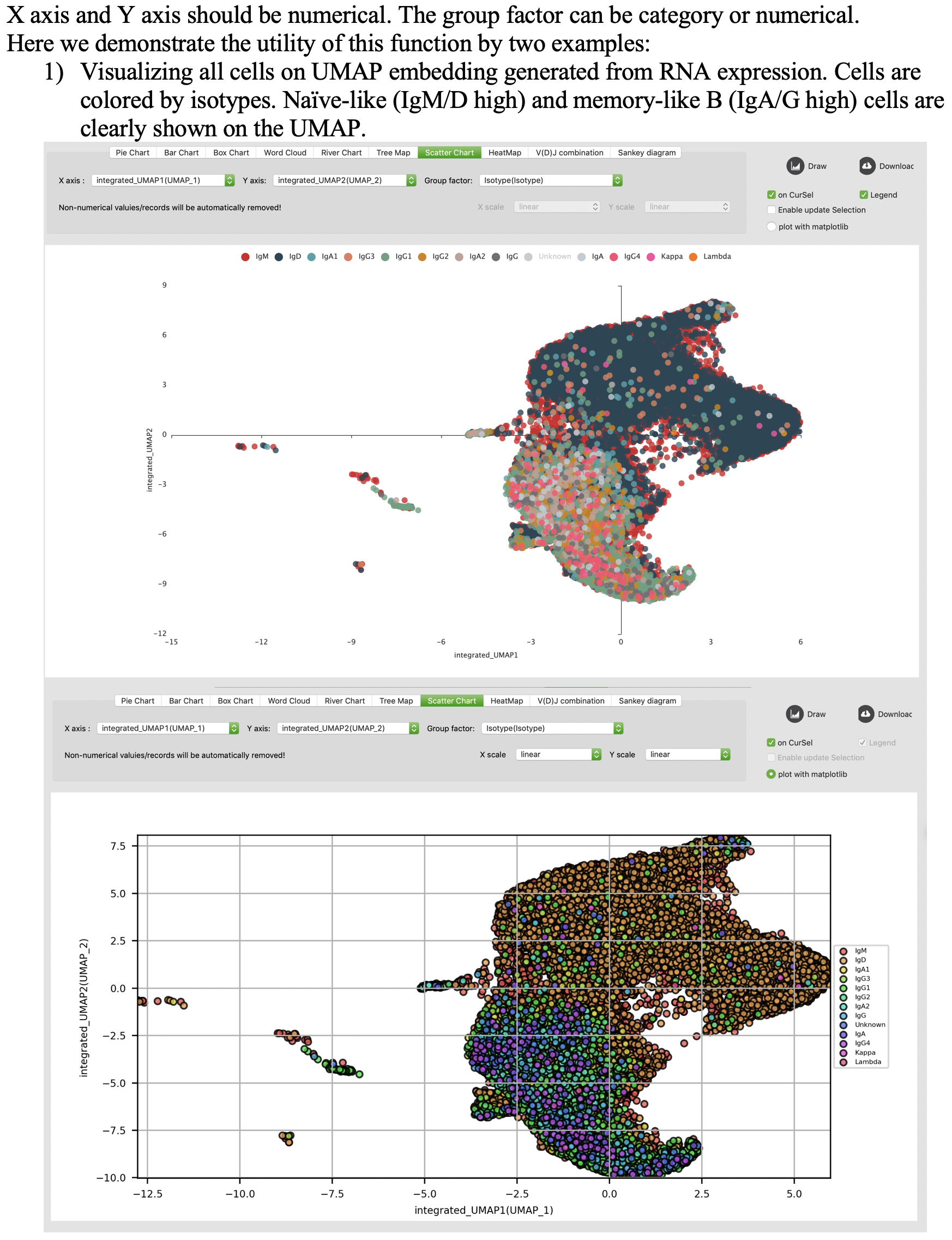
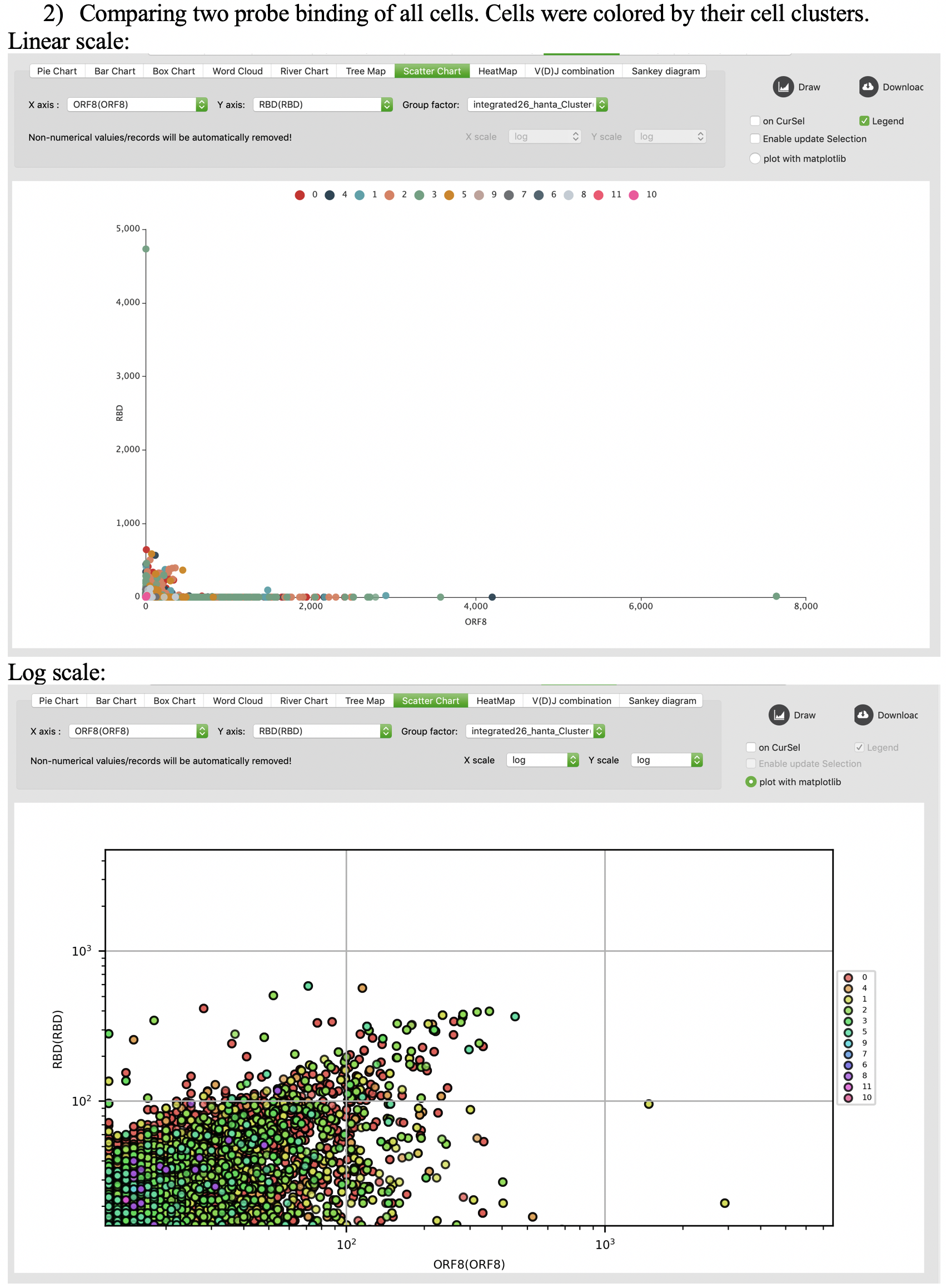
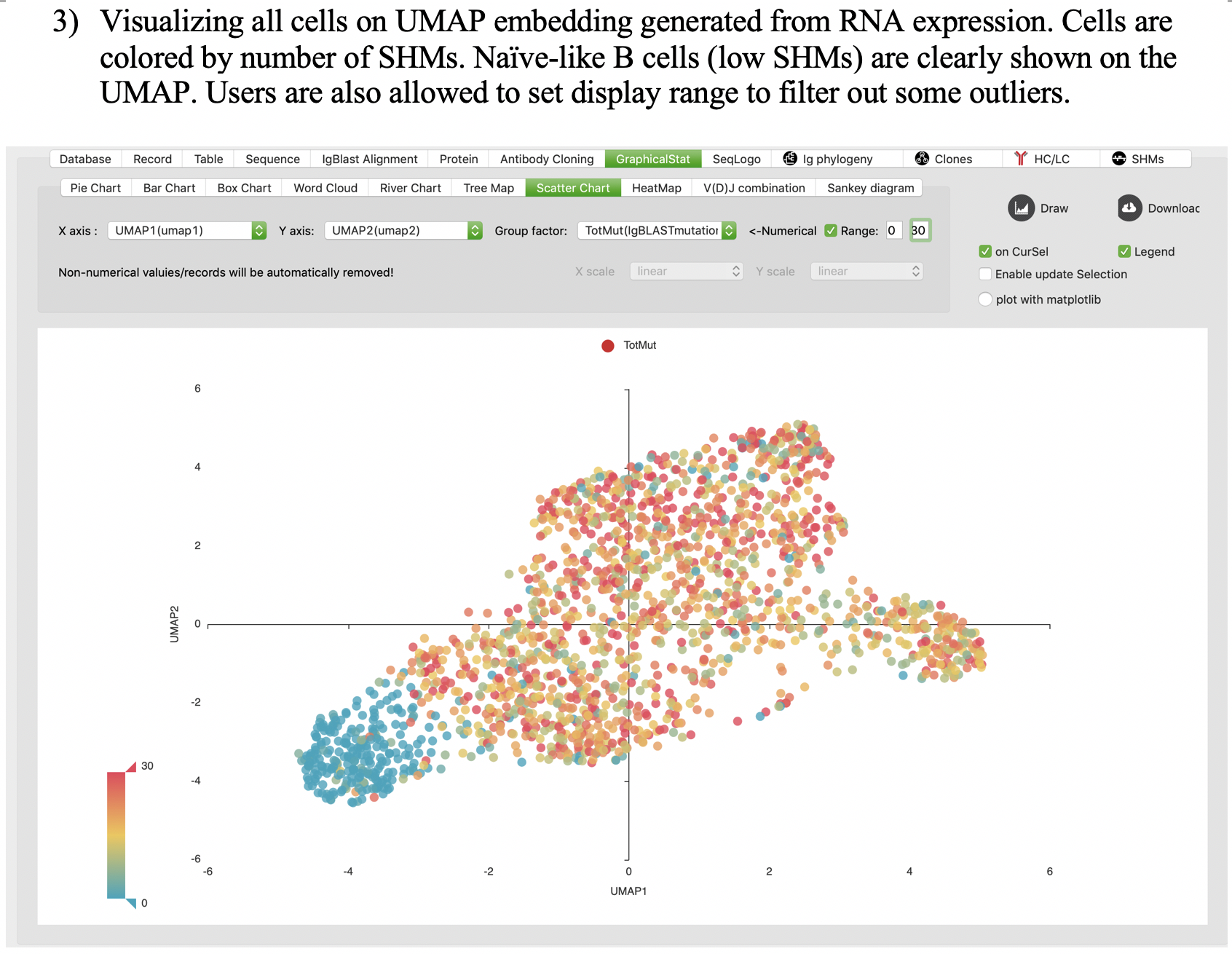
Heatmap
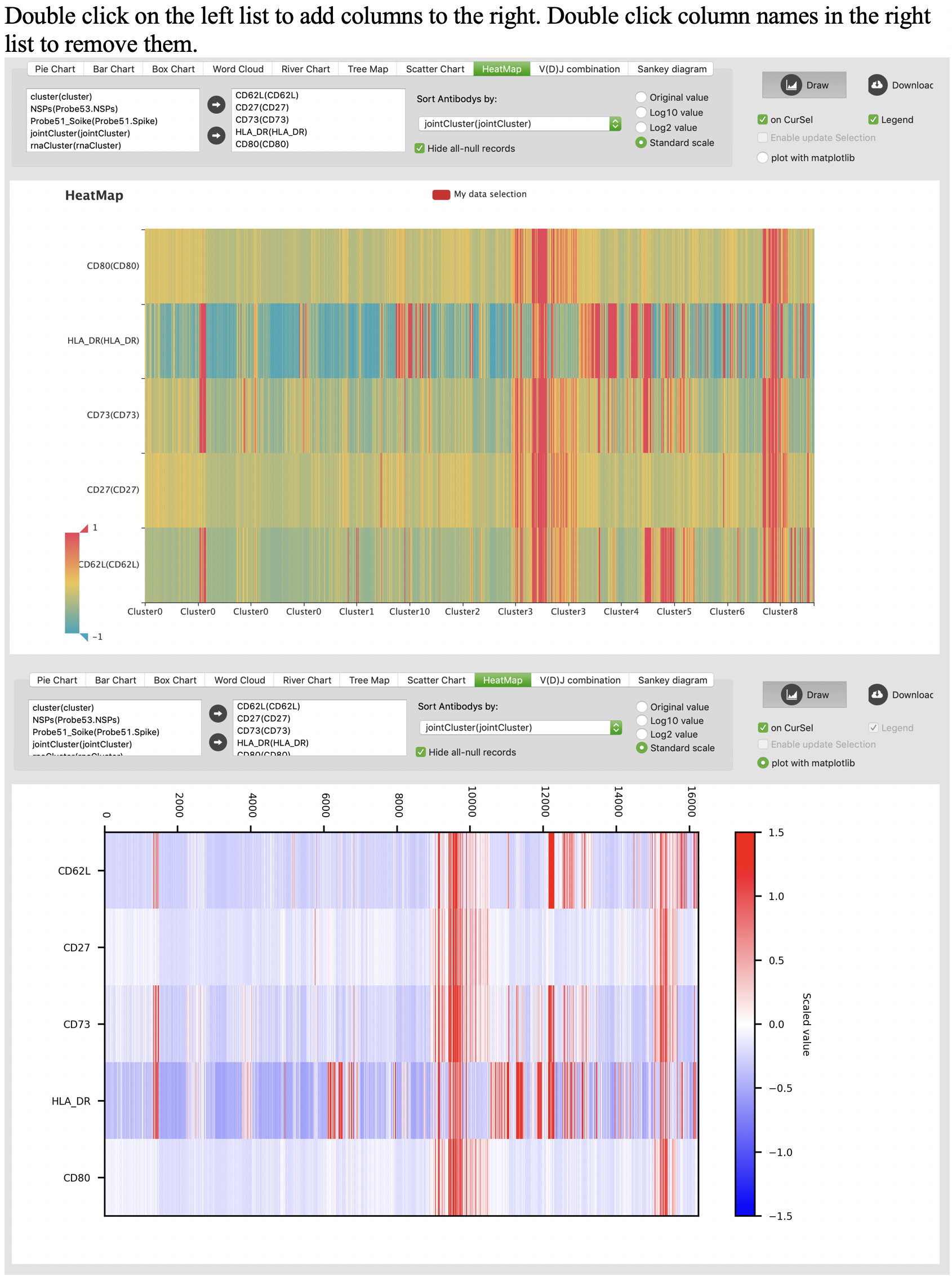
V(D)J combination heatmap
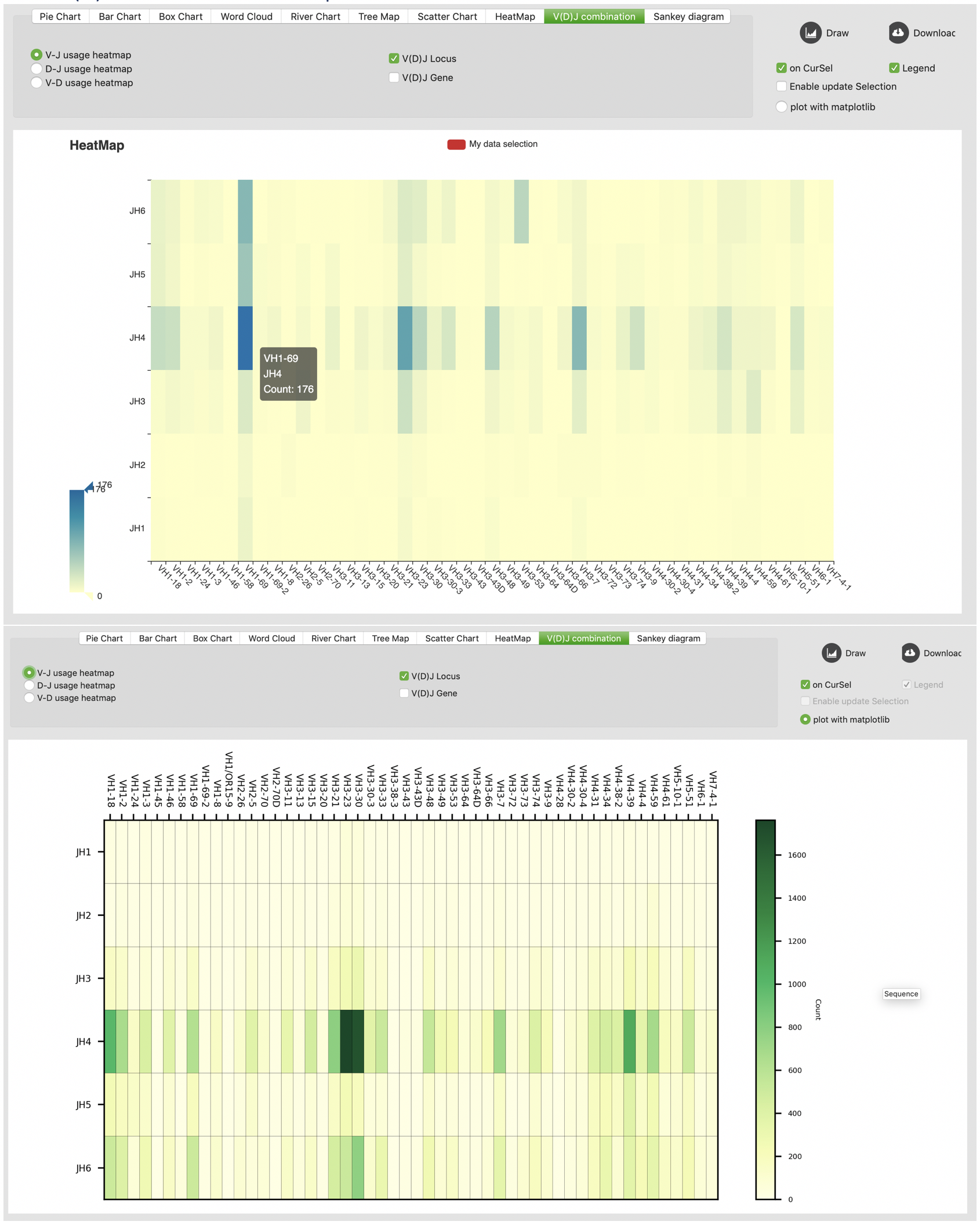
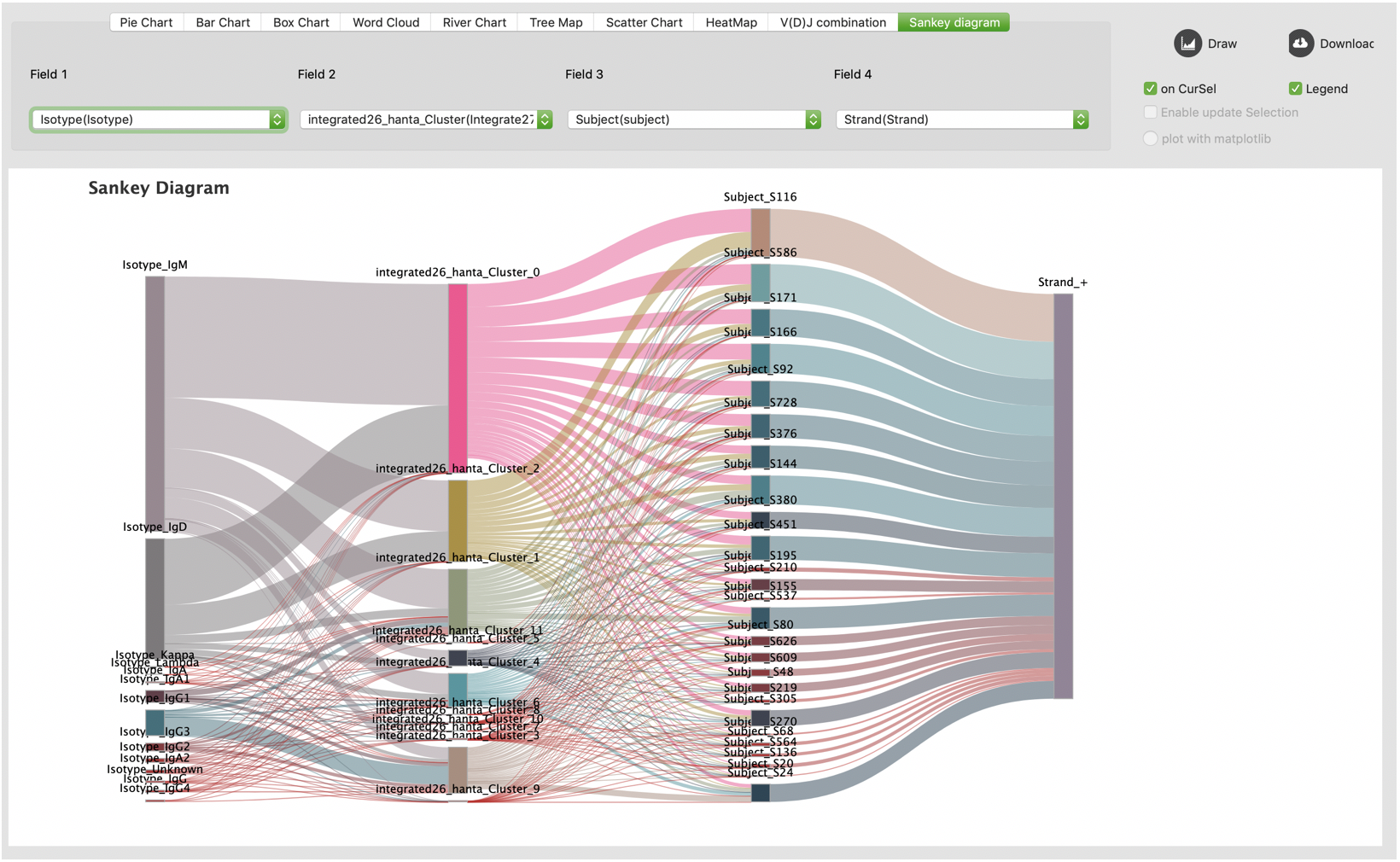
Sankey diagram
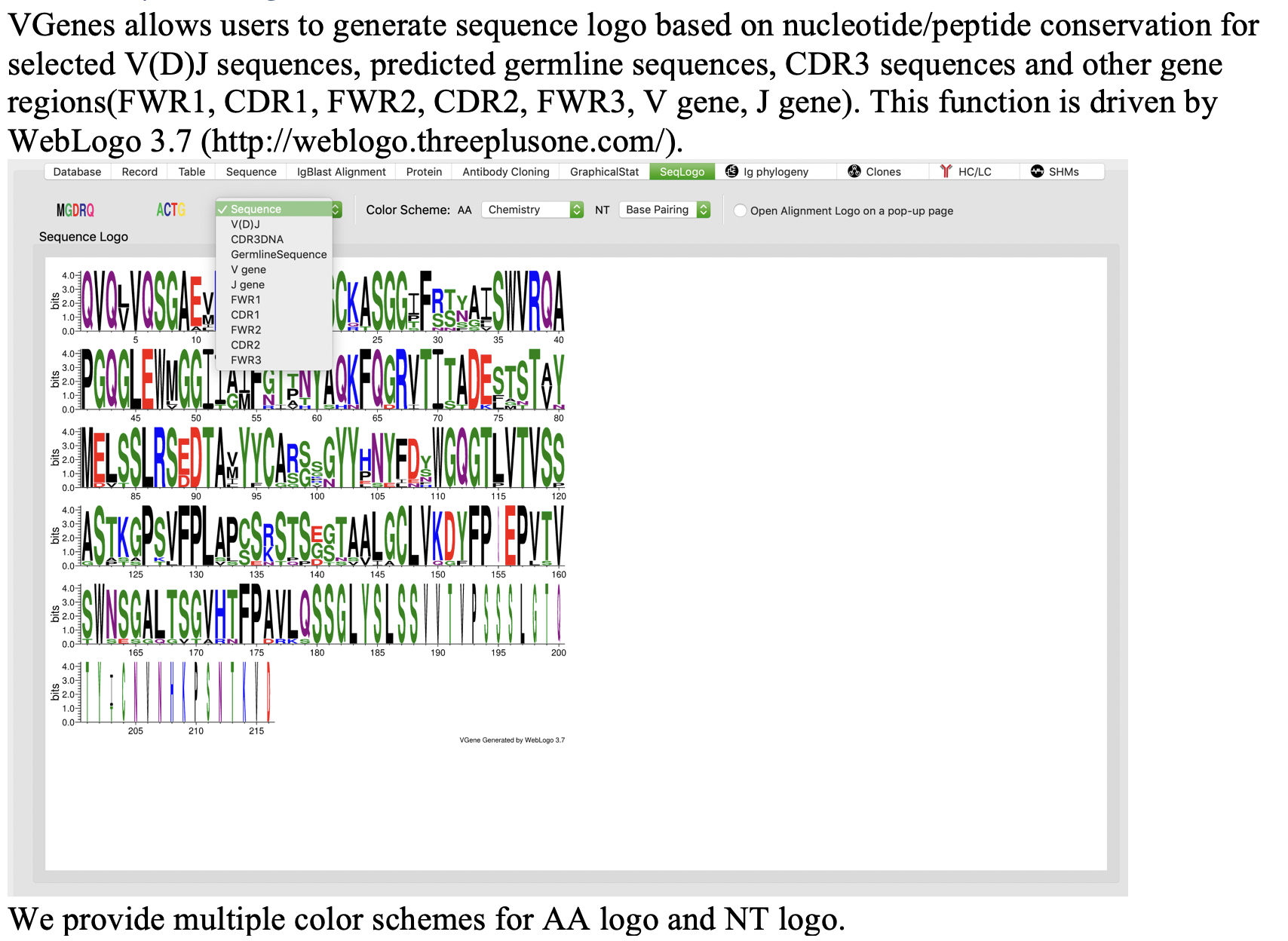
Sequence logo
-
Clonal analysis
VGenes helps users to identify B cell clones, perform phylogenic analysis and other clonal analysis.
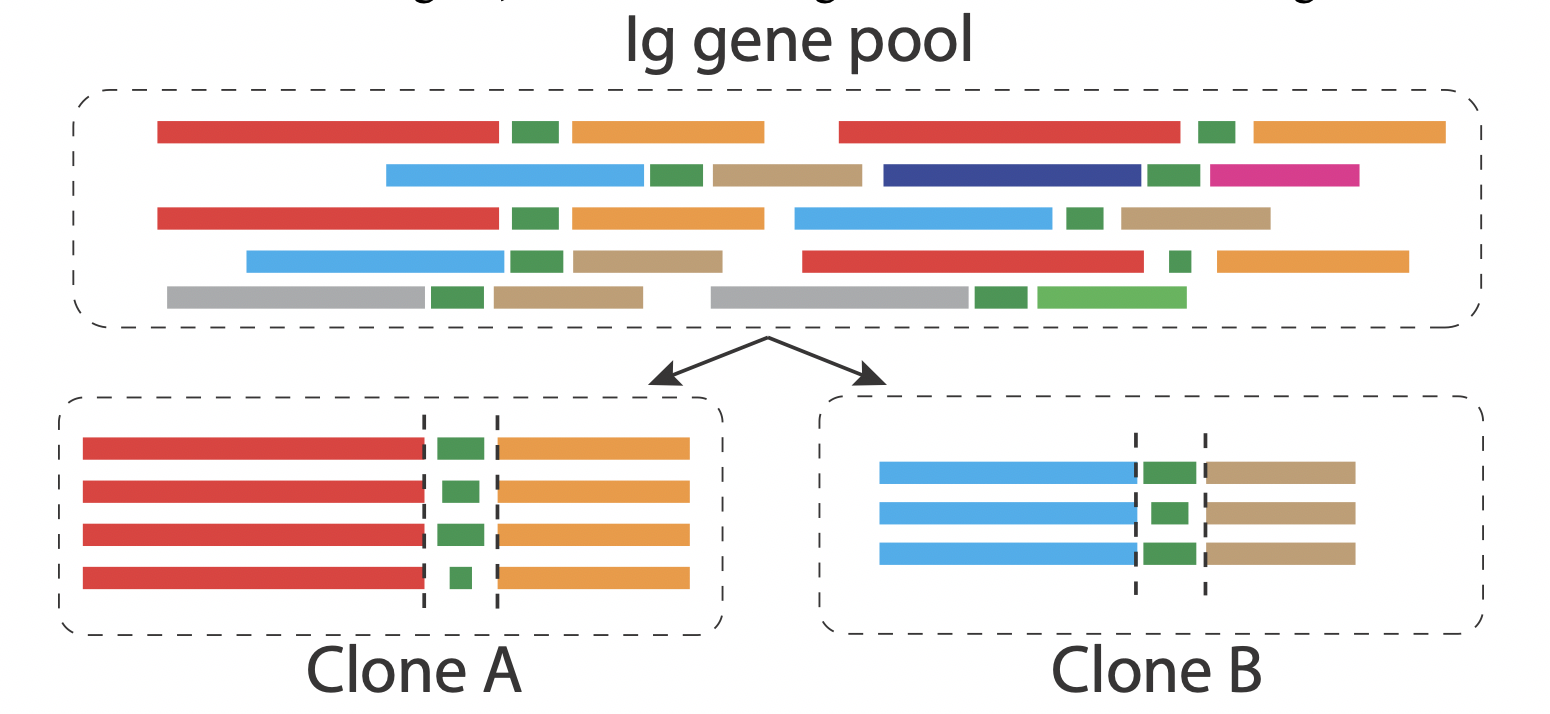
Identify clones
We developed function in VGenes to identify B cell clones. In brief, this clone classifier uses the following criteria to classify BCR sequences into different clones: members of a clone should share same IGHV and IGHJ gene, and their CDR3 gene should have same length. Users can active this function by clicking Tools->Find Clonal. Users can only check interested records, then the “Find clonal” functions will be only applied on those checked records.
Clone details
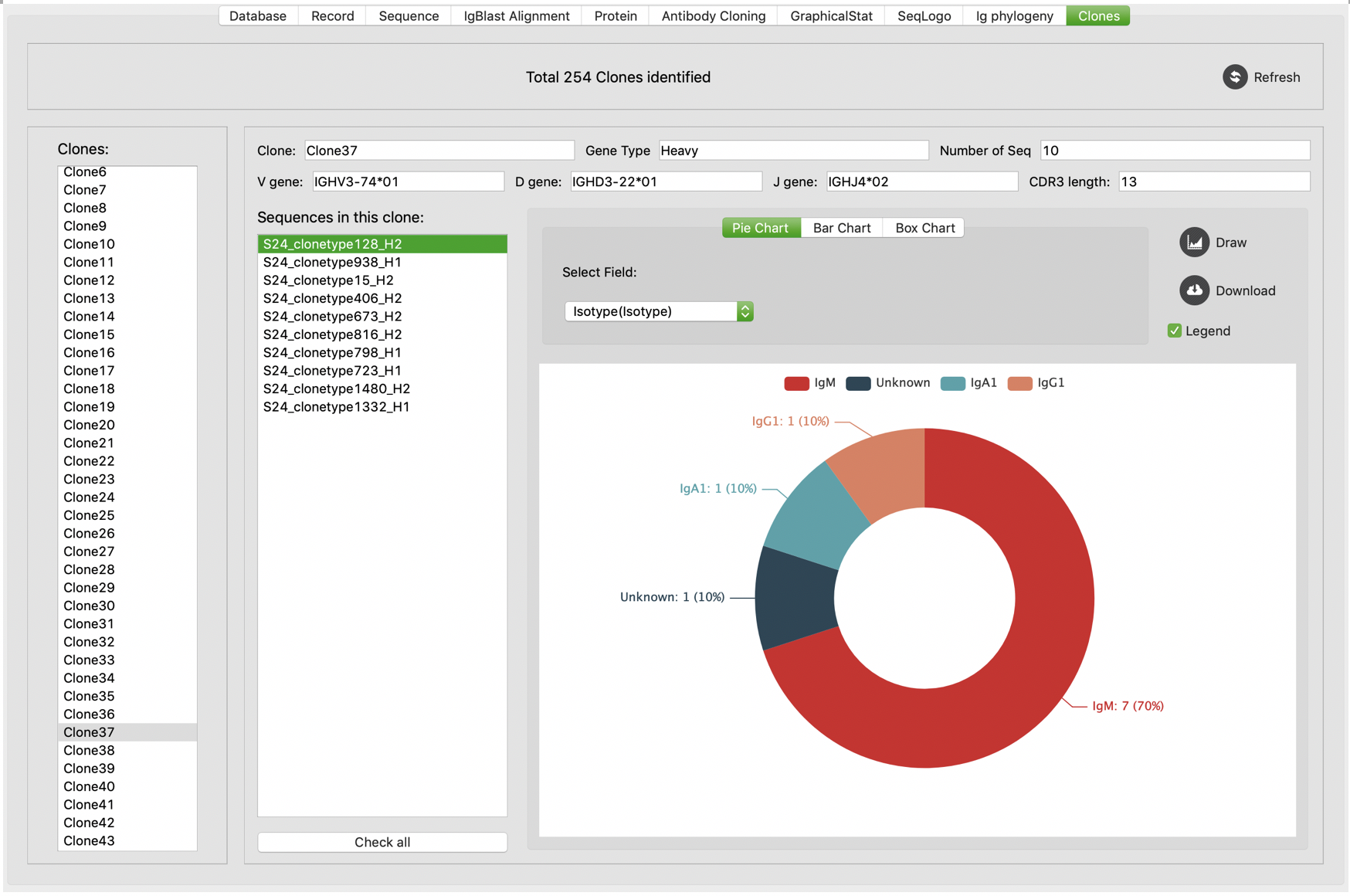
We also developed UI to display details of B cell clones. On “Clone” tab, all identified B cell clones are listed to the left, allowing users to select to show their details: clone members, V, D, J usage, CDR3 length, number of members, and gene types. Users are also allowed to do some graphical statistic within selected clone. Furthermore, users can click “check all” button to check all members of this clone for further analysis.
Identify potential public clones
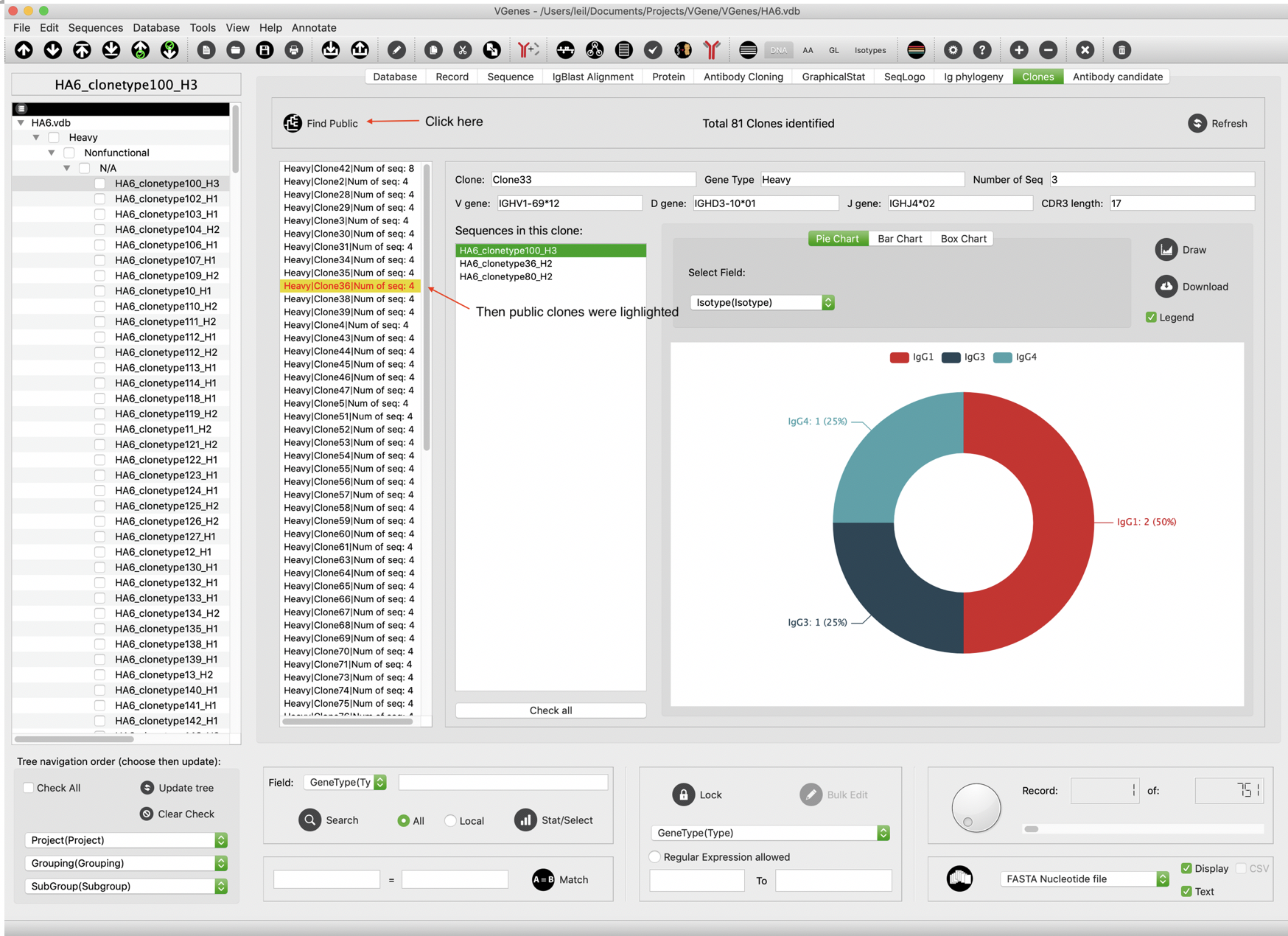
VGenes allows users to identify potential public clones by determining a field that contains subject/batch/dataset information. To active this function, users can click “Tools->IdentifyPublicClone” in menu, or just click “Find Public” button in “Clones” tab. As shown in the following figure, all potential “public clones” (clones that across multiple subjects/batches/datasets) were highlighted in red so that users can further check the details.
Ig phylogeny for clones
After B cell clones were identified, users are allowed to perform phylogenetic analysis for their interested clones. VGenes builds Ig phylogeny for selected clones using RAxML, a maximal likely-hood analysis tool (https://cme.h-its.org/exelixis/web/software/raxml/).
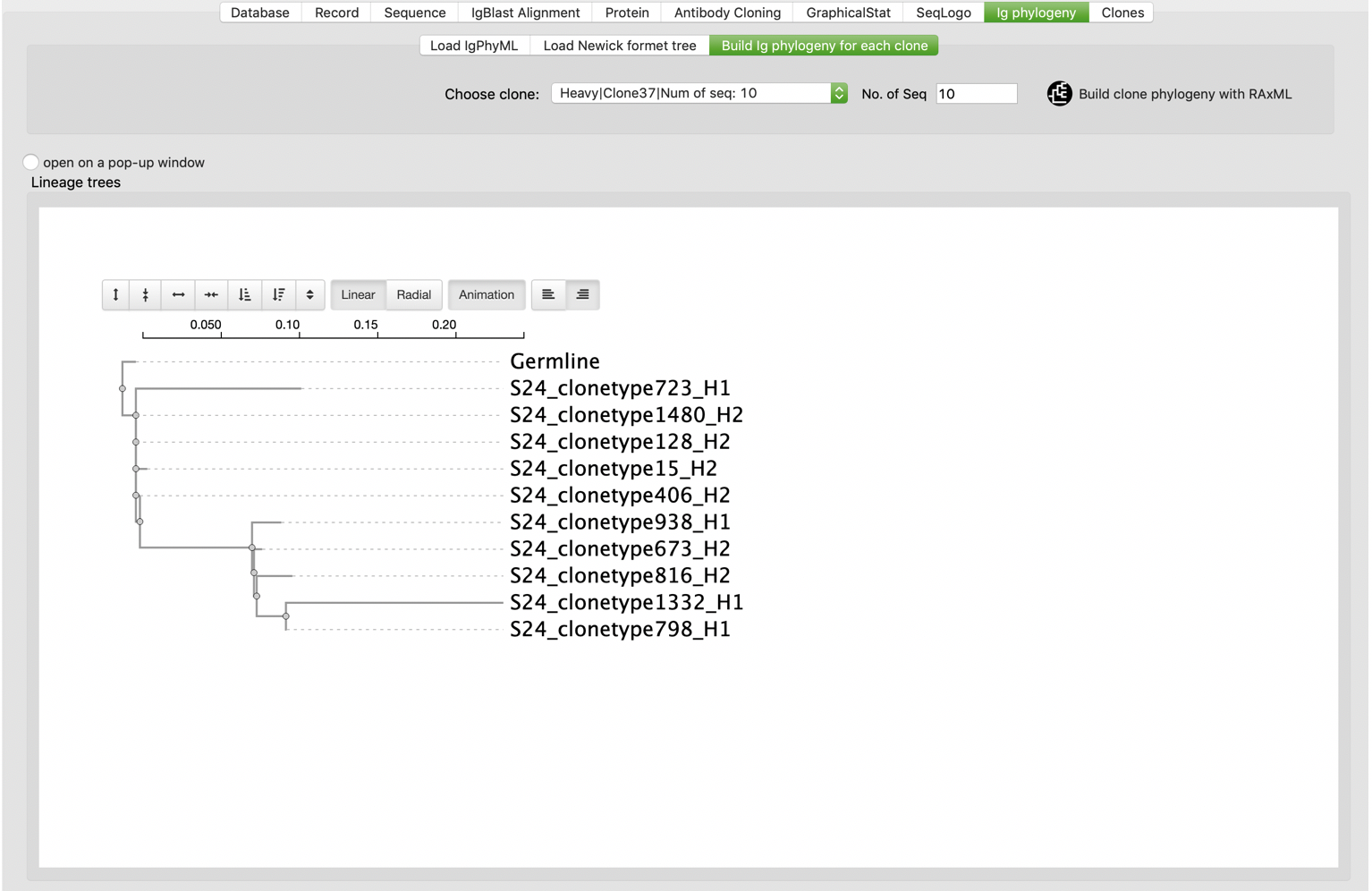
We also noticed that there are some Ig phylogeny specific tools available in research field, for example IgPhyML (http://evolve.zoo.ox.ac.uk/Evolve/IgPhyML.html). We developed functions to parse and visualize results from IgPhyML. Users should switch to “Load IgPhyML” tab under “Ig phylogeny” tab, then click “Load IgPhyML results” button to choose IgPhyML output file. Users also allowed to check “open on a pop-up window” to visualize trees on a pop-up window.
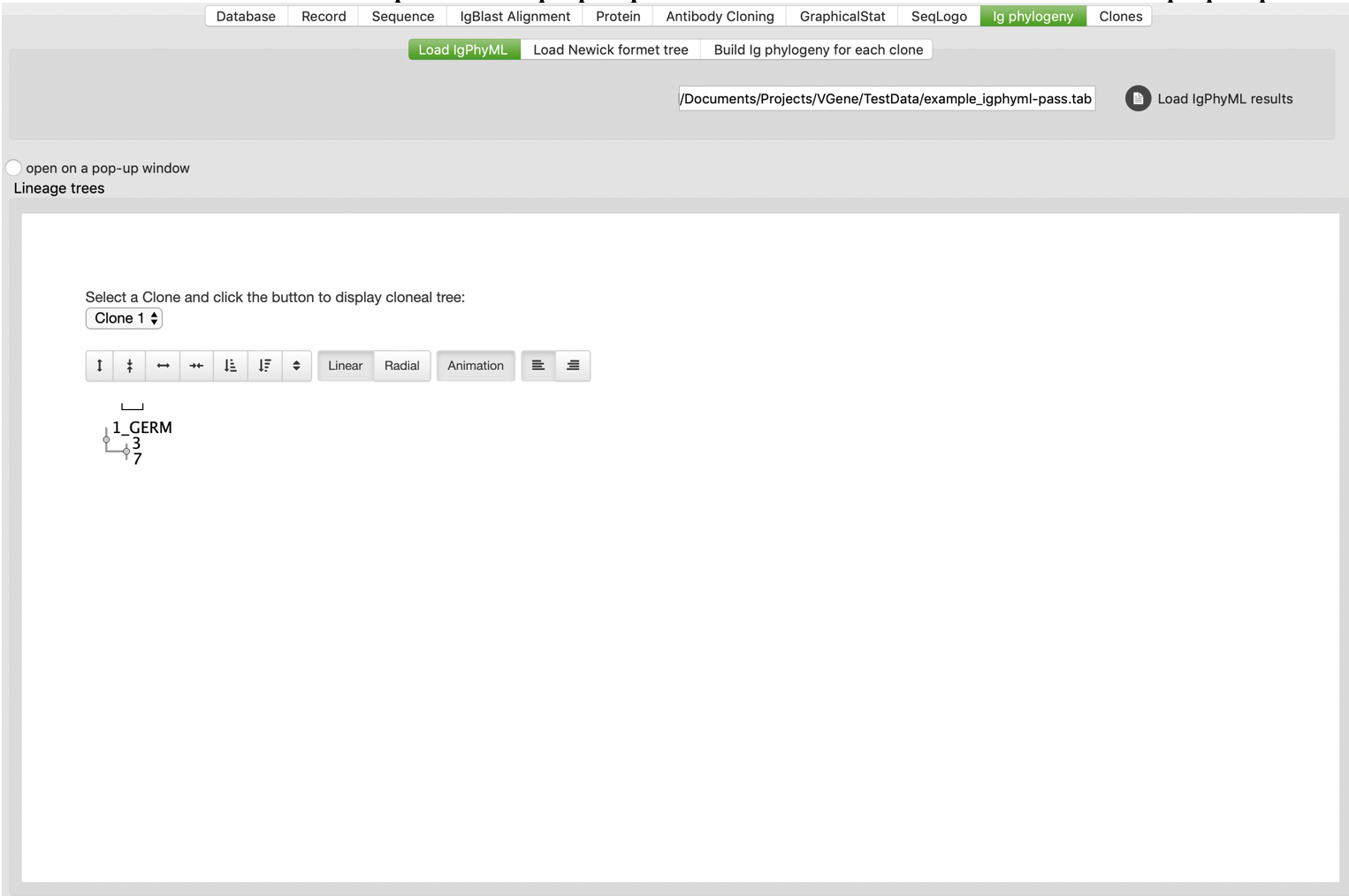
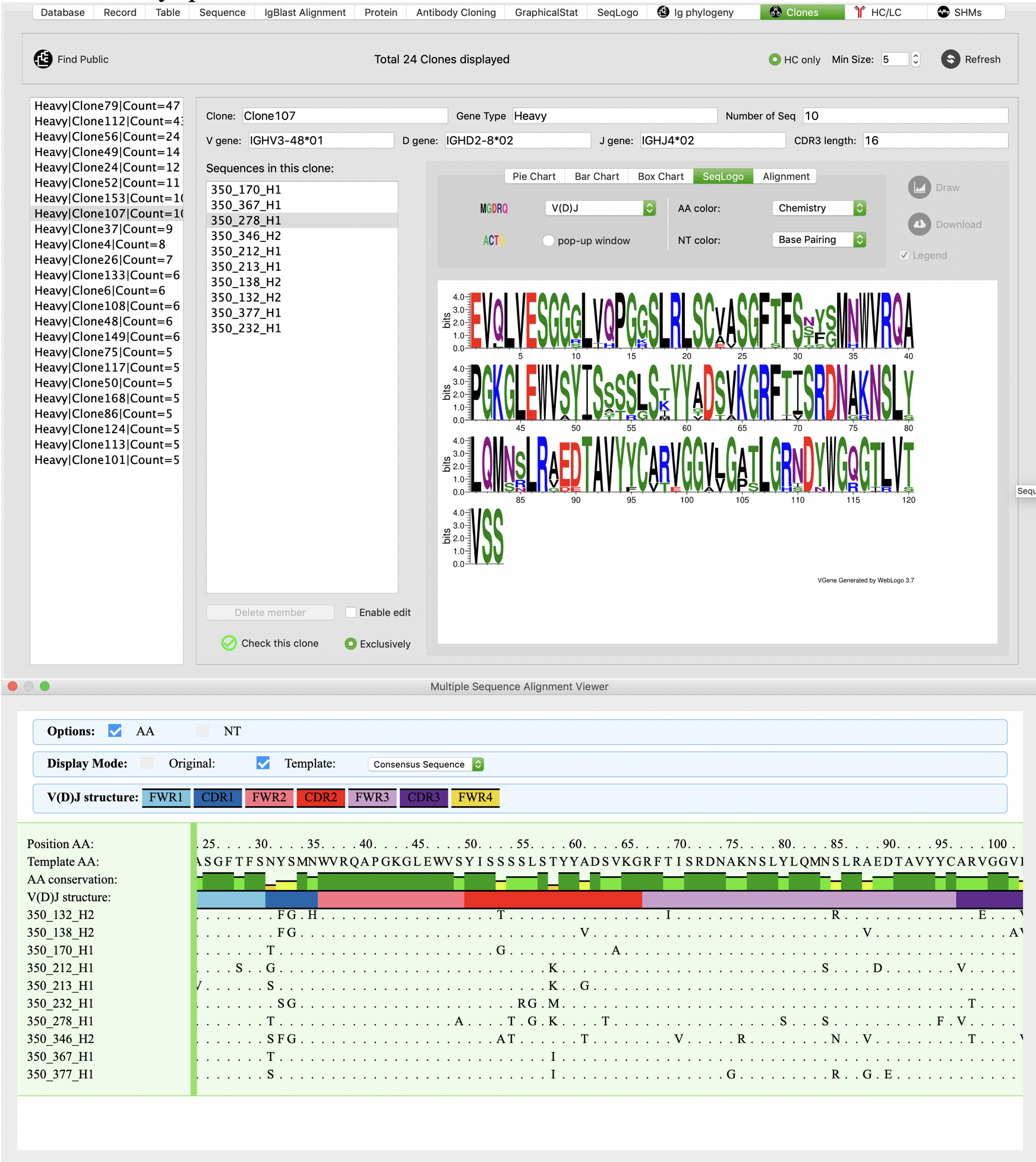
Sequence analysis for clones
We developed powerful functions for users to further investigate sequences and alignments of clone members. Users can easily access sequence logos and multiple sequence alignment of all members of any specific clone with a few click.
Investigation of both HC and LC of clones
-
Antibody mutational analysis
VGenes allows users to perform mutational analysis for their sequences
Mutational load analysis
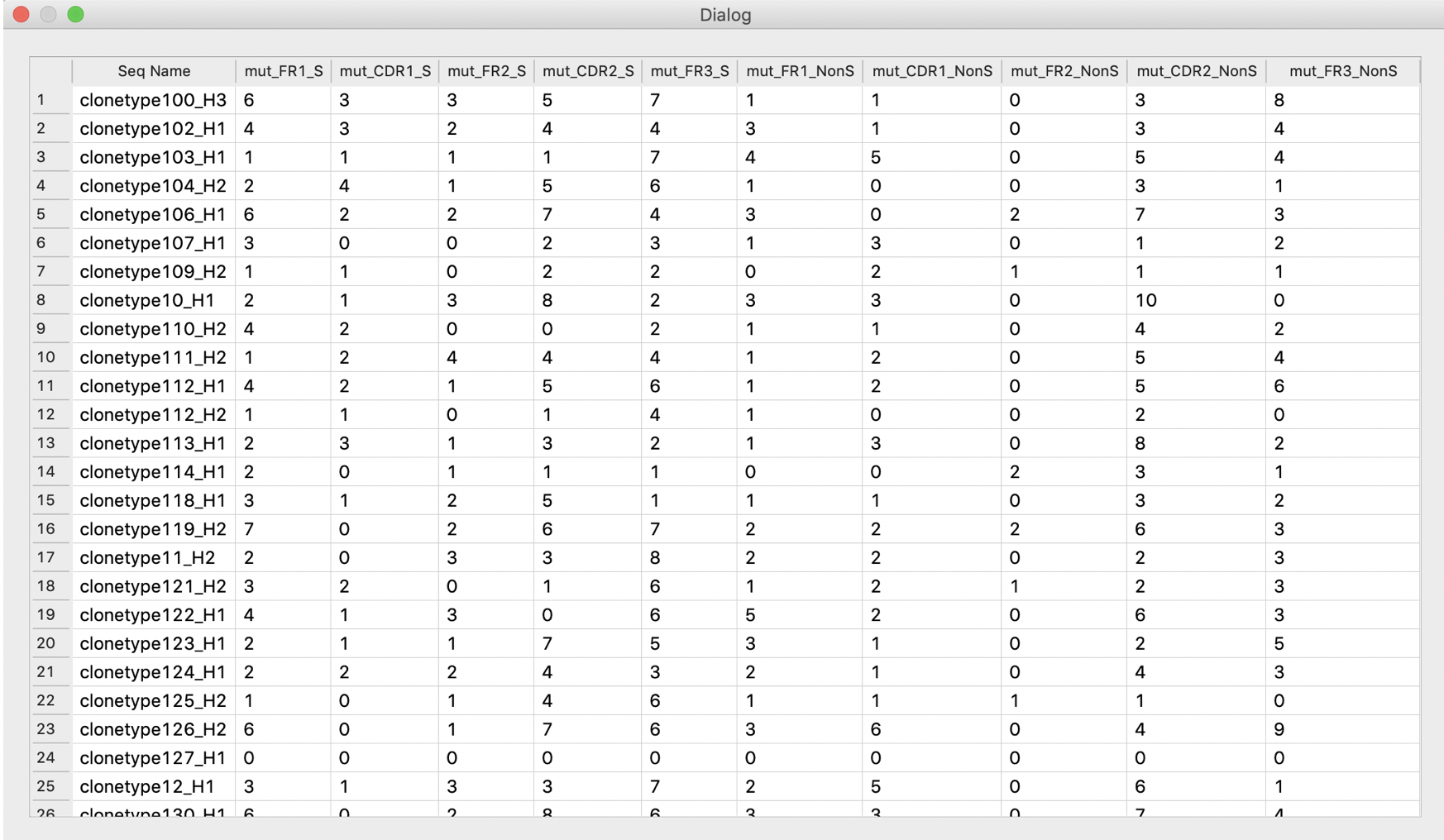
VGenes counts SHMs separately for each region of V genes, and allows users to perform statistical analysis on SHM of these regions.
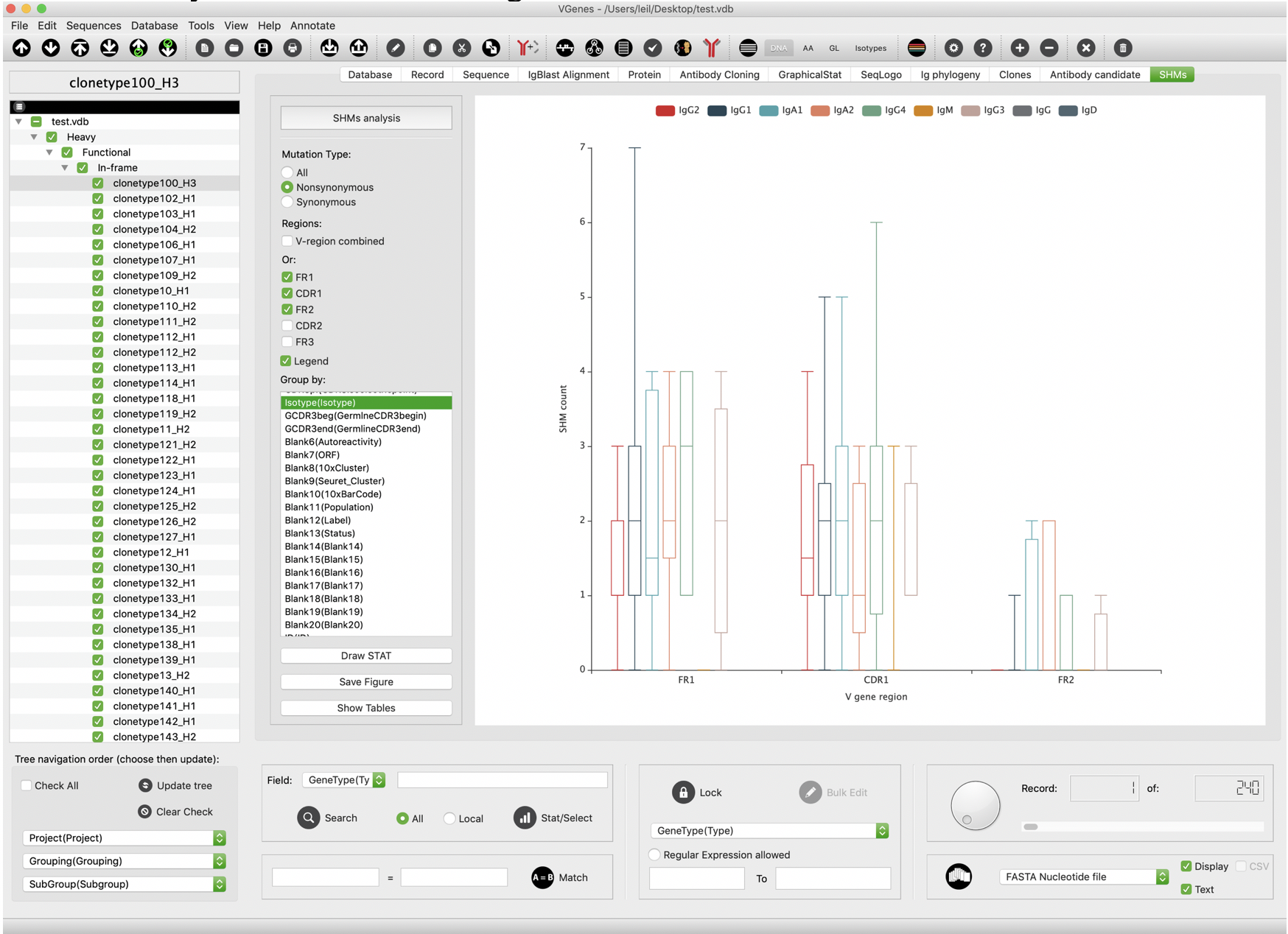
VGenes also can display all detail counts on a table.
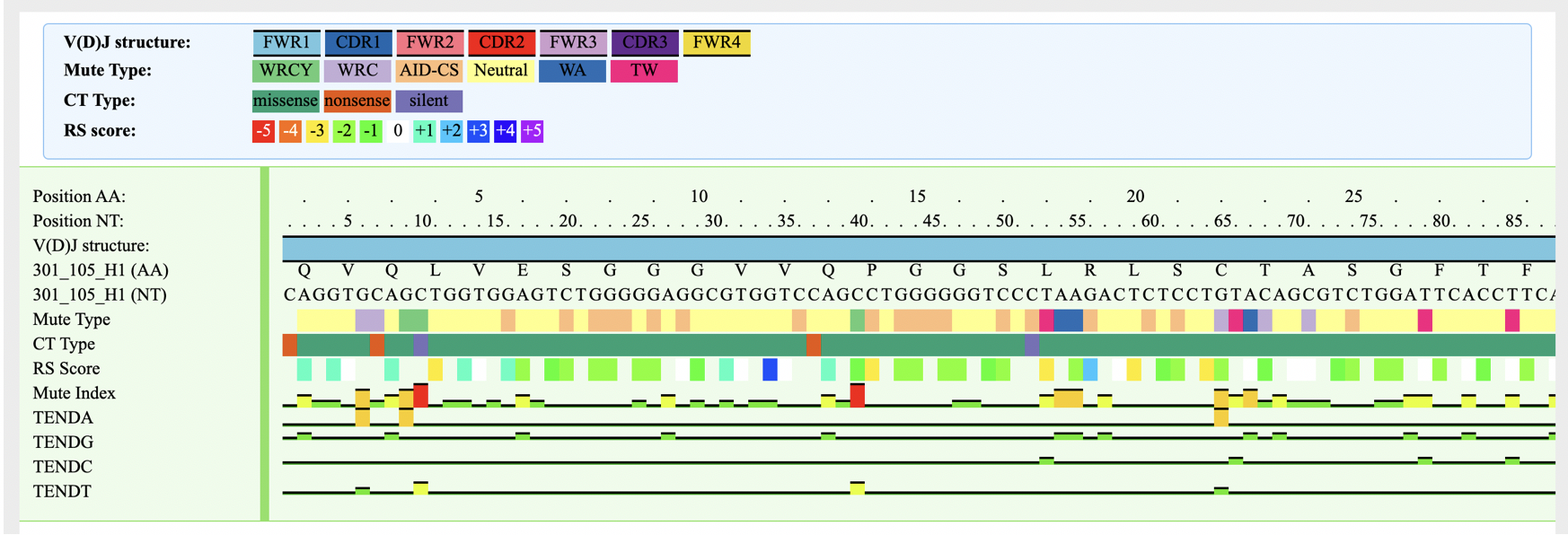
Inferring SHM targeting models (still under development)
VGenes also infers SHM targeting models for selected BCR sequences.
-
Antibody selection and cloning
VGenes designed a variety of functions to help users to select qualified antibody candidates efficiently and effectively. For conventional BCR datasets, VGenes allows users to select their interested subsets using any criteria or combination of multiple criteria, and then investigate their V(D)J combinations, CDR3 diversities, and clonal expansions to determine qualified antibody candidates for further experimental characterization. This workflow is particularly powerful and efficient by taking advantage of novel multimodal BCR datasets with antigen probes.
VGenes implemented a group of search and match functions designed for multimodal BCR data to facilitate users to focus on their interested subsets (e.g. antigen-specific B cells, B cells with high SHMs, naïve-like B cells) and explorer more about genetic preference and CDR3 diversities. Furthermore, we also developed an advanced graphical selector for users to apply multiple filtering criteria, allowing an effective selection of interested records from the multimodal datasets that have more than 100 different features. This selector is capable of visually selecting both numerical (numbers, e.g. number of SHMs, counts of antigen-probe binding, CDR3 length) and non-numerical (categories, factors, and levels, e.g. isotype, mRNA cluster, gene type) properties, allowing more flexibility and higher efficiency.
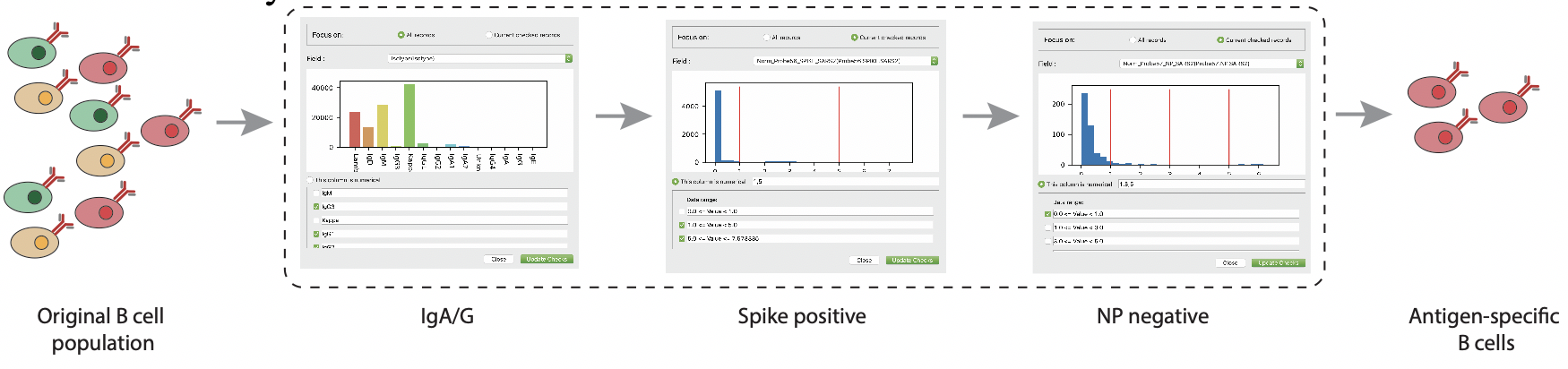
Antibody candidates selection
By adding antigen-probe binding to the searching criteria, VGenes helps users to rapidly select those candidates that most likely to be having high affinity and neutralizing. For example, using a combination of criteria of NP negative + Spike positive + IgA/G in a COVID-19 BCR dataset to select antibody candidates.
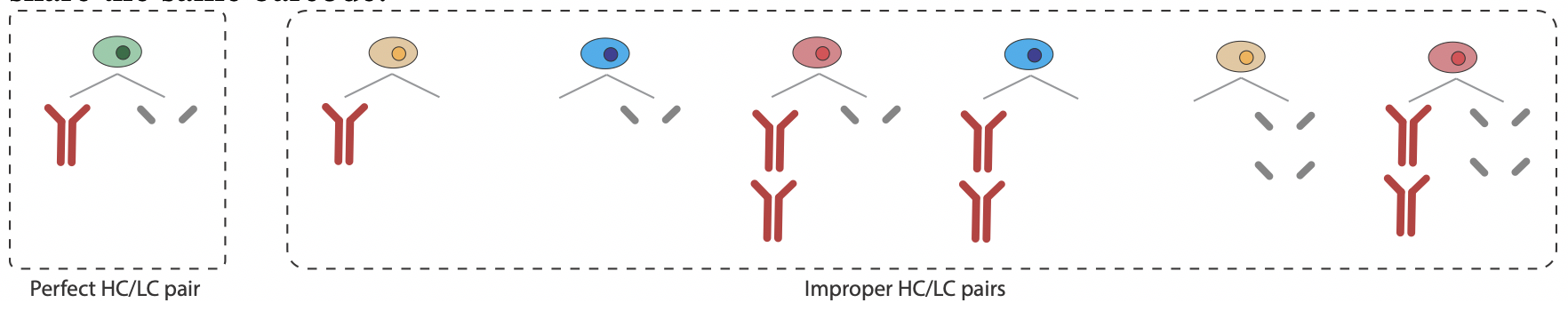
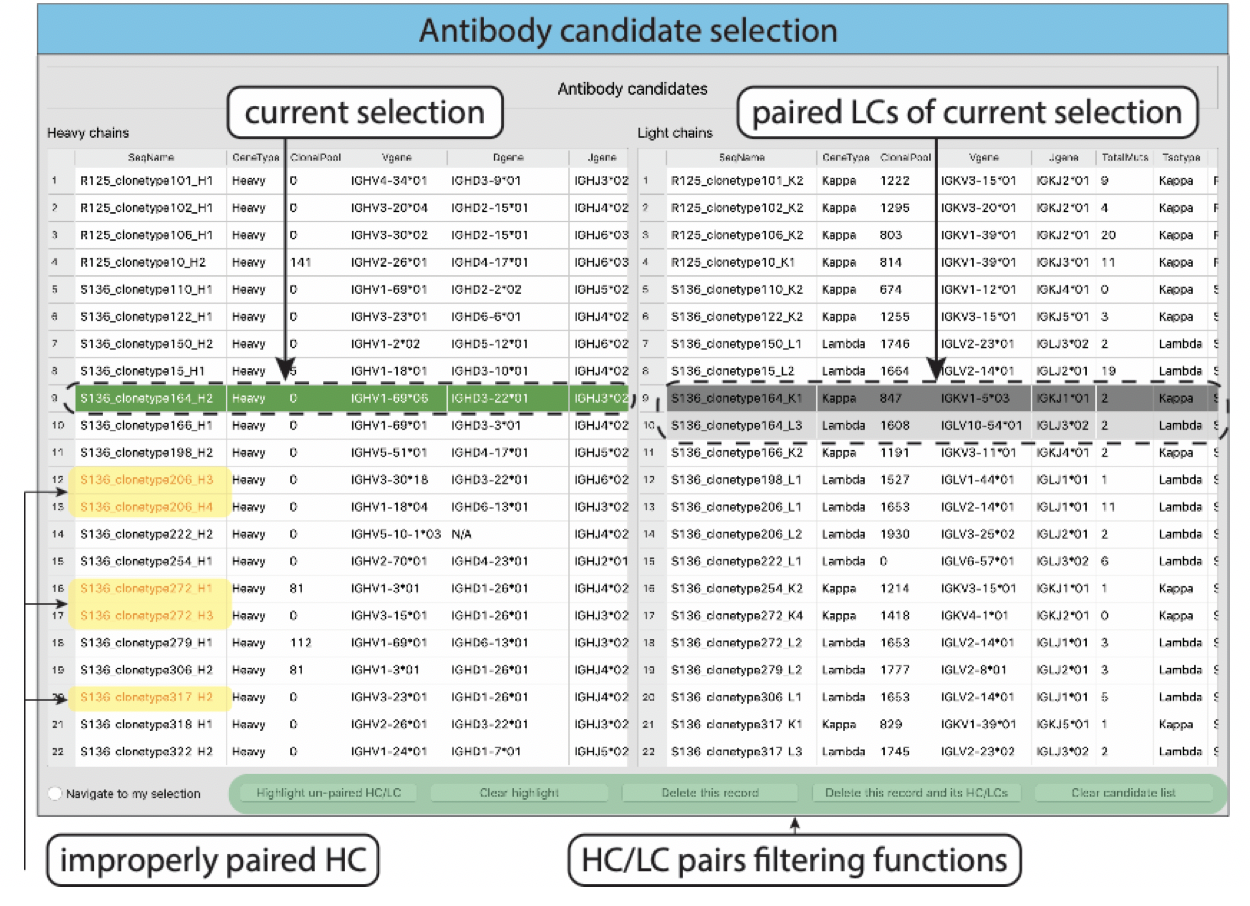
HC and LC pairing and cleaning
For single-cell datasets, after selected a subset of heavy chains, VGenes provides a function to help users to pair heavy chains (HC) with their light chains (LC) for the complete genome of antibodies. VGenes also helps users to easily check and filter out all improper B cell records (e.g. unpaired HC and LC, doublets, and multiplets) by investigating the number of HC and LC that share the same barcode.
Sampling
VGenes integrated three sampling methods for both bulk BCR data (sampling from BCR sequences, usually heavy chain) and single-cell BCR data (sampling from HC and LC pairs). VGenes allows users to determine all candidates by either checking all individual sequences or pairing HCs and LCs for complete antibody genome. After users determined candidates for sampling, three sampling methods, random sampling, stratified sampling, and representative sampling, are implemented for users to choose according to their requirement. In random sampling, VGenes randomly pick a user-specified number of samples. In stratified sampling, users are allowed to determine a group factor, then randomly sample from each group of the group factor. VGenes allows users to set a fixed or scaled sample size for each group. In representative sampling, we adopted an sampling algorithm from a previous study that can select representative samples by balancing multiple factors. This algorithm has three key steps: vectorizing, distancing, and clustering. Vectorizing step collects values of selected factors for each sample, and packs them into a vector. This step also normalize different factors to balance the contribution of each factor, avoiding the bias due to differences in magnitudes of measurements. The distancing step calculates pairwise numerical distances from the vectors. As there are two types of factors in VGenes, numerical factor and none-numerical factor, we designed different models to quantify differences of different factors. In brief, for numeric factors, the difference between two values is considered as their difference. For none-numeric factors, a binary system was applied: 0 indicates no difference and 1 indicates difference. Then the distance between any two sample will be calculated using a L2 norm (Euclidian distance) from the vector of difference. Using the pairwise distance matrix, clustering step clusters all samples into a user-specified number of clusters using K-medoids algorithm. Then VGenes considers all clusters centers as selected samples. VGenes also allows users to determine a few important factors that need to be comprehensively sampled (100% coverage requirement). For details about this representative sampling workflow please refer to a previous study.

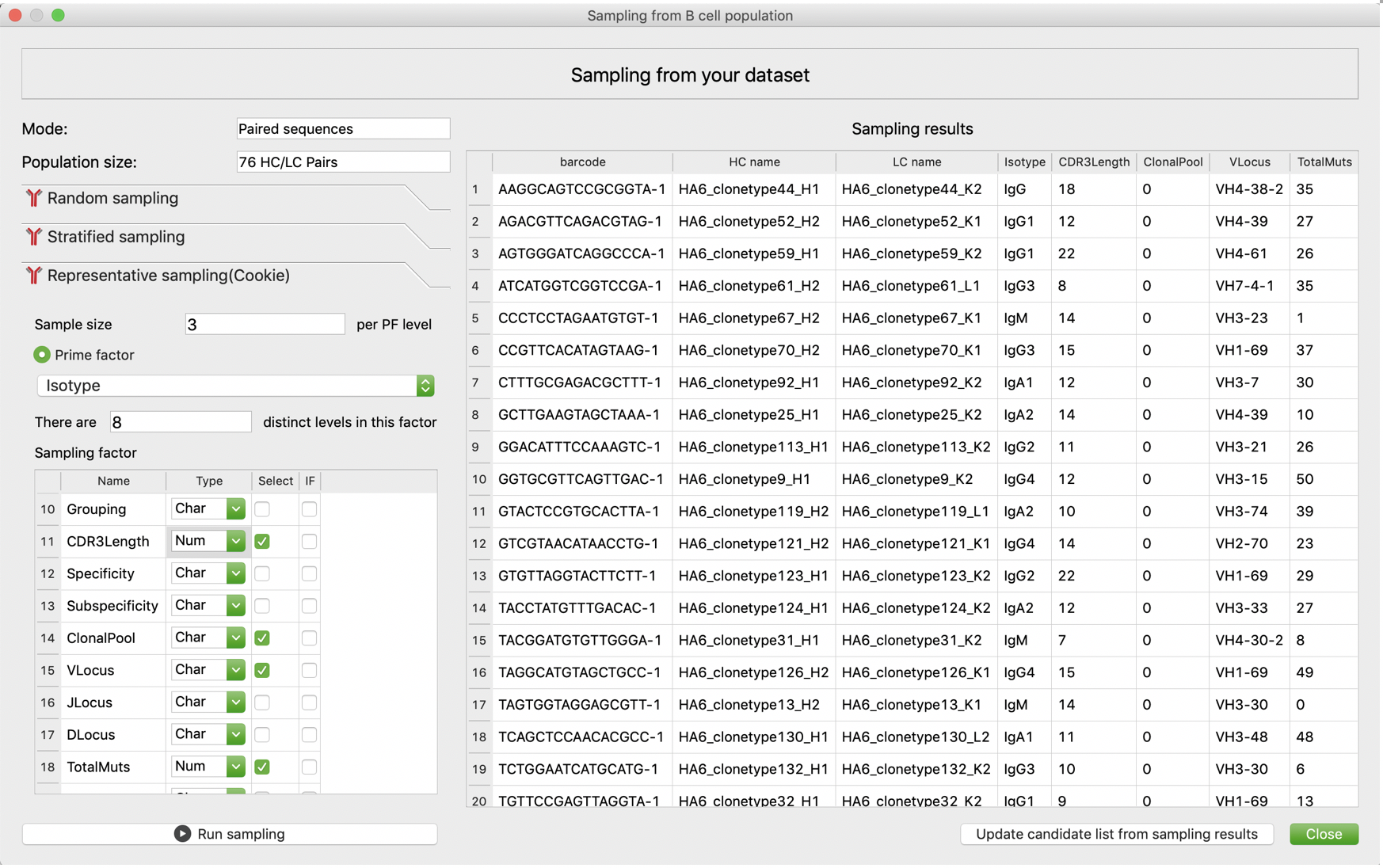
After sampling, users are allowed to click “Update candidate list from sampling results” (paired HC/LC mode) or “Select my sampling results in VGenes” (individual sequence mode) to apply sampling results to the candidate list or tree selection for further steps.
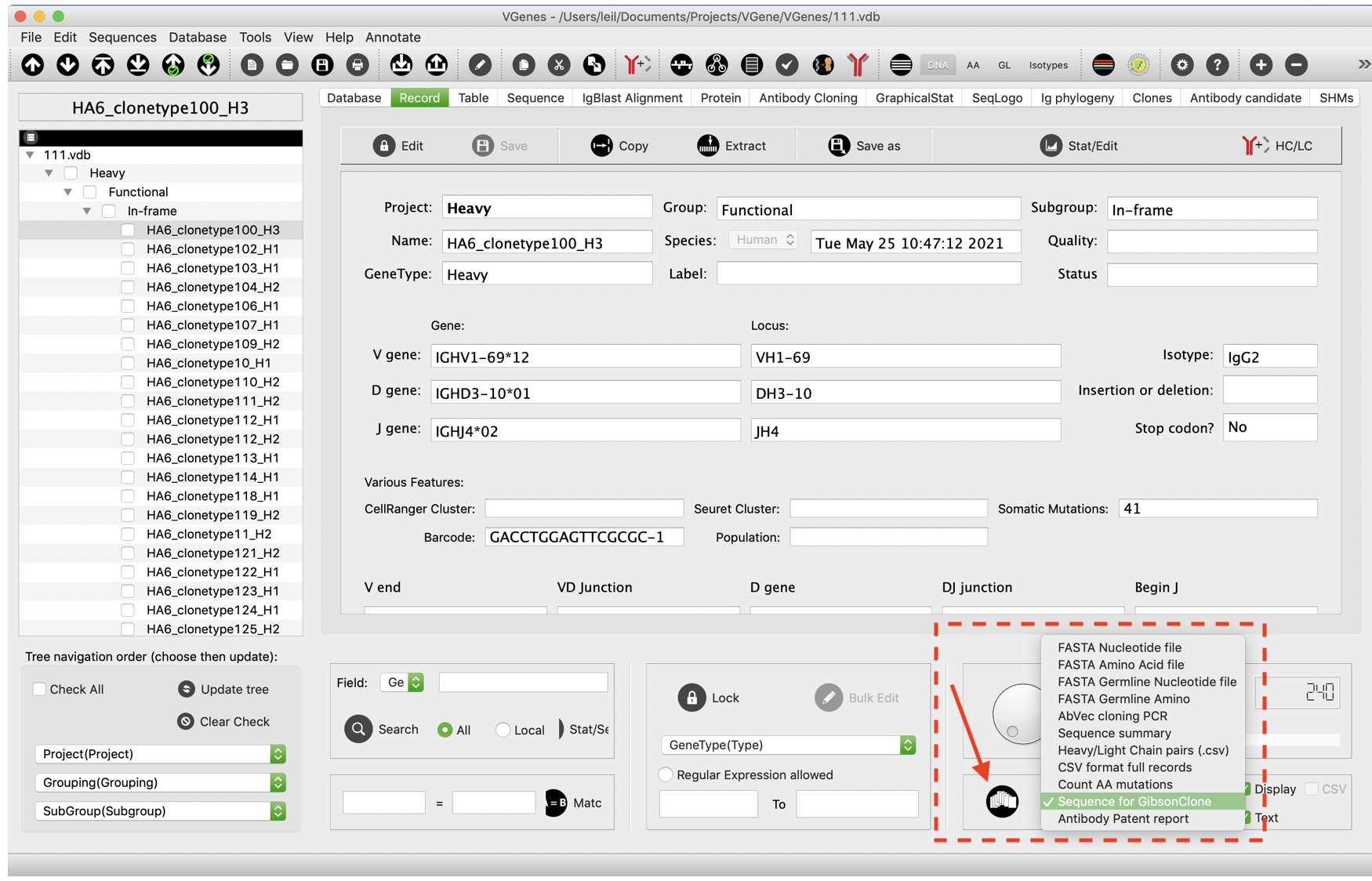
Antibody cloning
For paired HC/LC antibodies, users are allowed to pair them and check the sequence in a candidate list table.
Users can choose “Sequence for Gibson Clone” from right bottom corner and click the button to the left to open the dialog for antibody sequences generation. This function will prioritizes paired HC/LC antibodies in the HC/LC page. If there is no sequences in the candidate list, this function will try to pull all checked sequences.
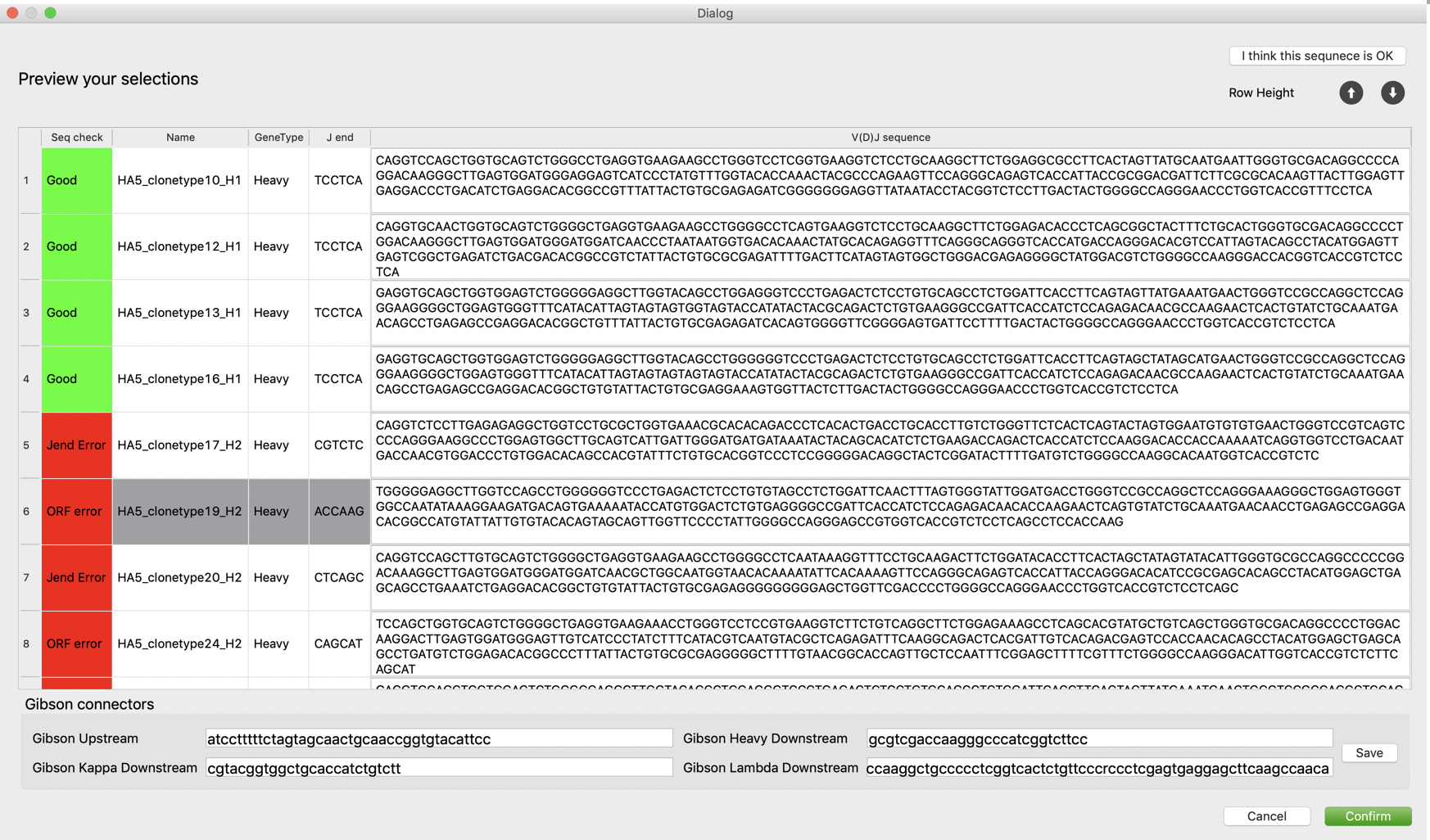
While sequences are loaded, VGenes checks all details of BCR sequences, including J-end sequence, translated sequences, and open reading frame, and highlights all issues with a red flag. Users are allowed to manually edit their sequences, and VGenes will automatically re-check users’ edits and turn the flag to green if the inspection passes.
There are two common red flags: “Jend error” and “ORF error”. “Jend Errors” means the end of J gene is not correct, usually because of 1 or 2 extra nucleotides of the V(D)J sequence, users can just simply delete them. The translated AA, Jend info and the sequence check flag will be automatically updated. For Heavy chain, we define a 6-mer pattern “TCCTCA” as acceptable J-end and will highlight all different J-ends to notice users for a further check. For light chains, we define 6-mer patterns “TCGAAC”, “ATTAAA”, “ATCAAA” for kappa chain and define a 6-mer pattern “GTCCTA” for lambda chain as acceptable J-end. Notably, synonymous mutations were observed in the last six nucleotides of BCR genes, for example, “TCATCA” or “TCCTCG” instead of “TCCTCA” for heavy chains. For those cases, VGenes will highlight them and allows users to determine to whether accept or exclude them. Users can click "I think this sequence is OK" button on the top to make the flag green manually if they think the sequence is OK. “ORF error” means VGenes found something wrong in the translated AA sequence, so we suggest users check if there is any ambitious nucleotide in your sequence, or the beginning of VDJ sequence missed 1 or 2 nucleotides so that the ORF is wrong. The UI of Gibcon clone page is linked with the main VGenes page, therefore selections on main page will be automatically updated when users click a record on Gibson clone page. This feature is designed to facilitate users to check the sequence details for solving “ORF error” issue. For “ORF error” issues, users can click and these records and investigate their IgBlast alignment on the main page to figure out if they need to fill the missed 1 or 2 (or delete the extra 1 or 2) nucleotides in the beginning of sequence to make sure the V(D)J sequence is correctly translated.
Users are also allowed to manually turn the flag of a specific sequence to green if they are confident with the current sequence. After the sequence inspection, users should provide Gibson upstream and downstream connector sequences. VGenes currently supports three expression vectors: Gibson AbVec, HT-AbVec, and AbVec classic. The connector sequences of these vectors are also customizable for better compatibility with users’ own cloning systems. Users can directly edit on the text editors to apply their own sequences for vectors. Users can click “Save” button to save the customized setting for later use.
After users click “Confirm”, VGenes will generated sequences for all records with indicator “Good” in a CSV file or FASTA file.

VGenes automatically added 5’ and 3’ connector sequences to all BCR sequences. All nucleotides of connector sequences are in lower case and all nucleotides of BCR sequences are in upper case. As shown in the following example:
>HA5_48_H1 atcctttttctagtagcaactgcaaccggtgtacattccCAGATCCAGCTGGTGCAGTCTGGGGCTGAG ... AGGGAACCCTGGTCACCGTCTCCTCAgcgtcgaccaagggcccatcggtcttcc
Export data
-
FASTA sequences
Users are allowed to export FASTA sequences for selected records. Users can export: 1) nucleotide sequences 2) amino acid sequences 3) nucleotide germline sequences 4) amino acid germline sequences
-
AbVec cloning PCR report
Cloning report for AbVec cloning PCR primers for selected sequences.
-
Sequence summary report
A summarized report for selected sequences. It includs the following key information:Name, Project, Vgene, Dgene, Jgene, Vlocus, Jlocus, Productivity, IgBLASTmutationcount, CDR3DNA, CDR3peptide, CDR3length, CDR3isoelectricpoint, ClonalPool, Isotype, Sequence, SharedName.
-
Heavy/Light chain pairs report
This function will match paired heavy chain (HC) and light chain (LC) according to their barcode information for selected records. Only HC/LC pairs have 1 HC and 1 LC will be exported. HC/LC pairs have multiple HCs or LCs will be excluded.
-
CSV format full records
This function will export all detailed information for selected records in CSV file.
-
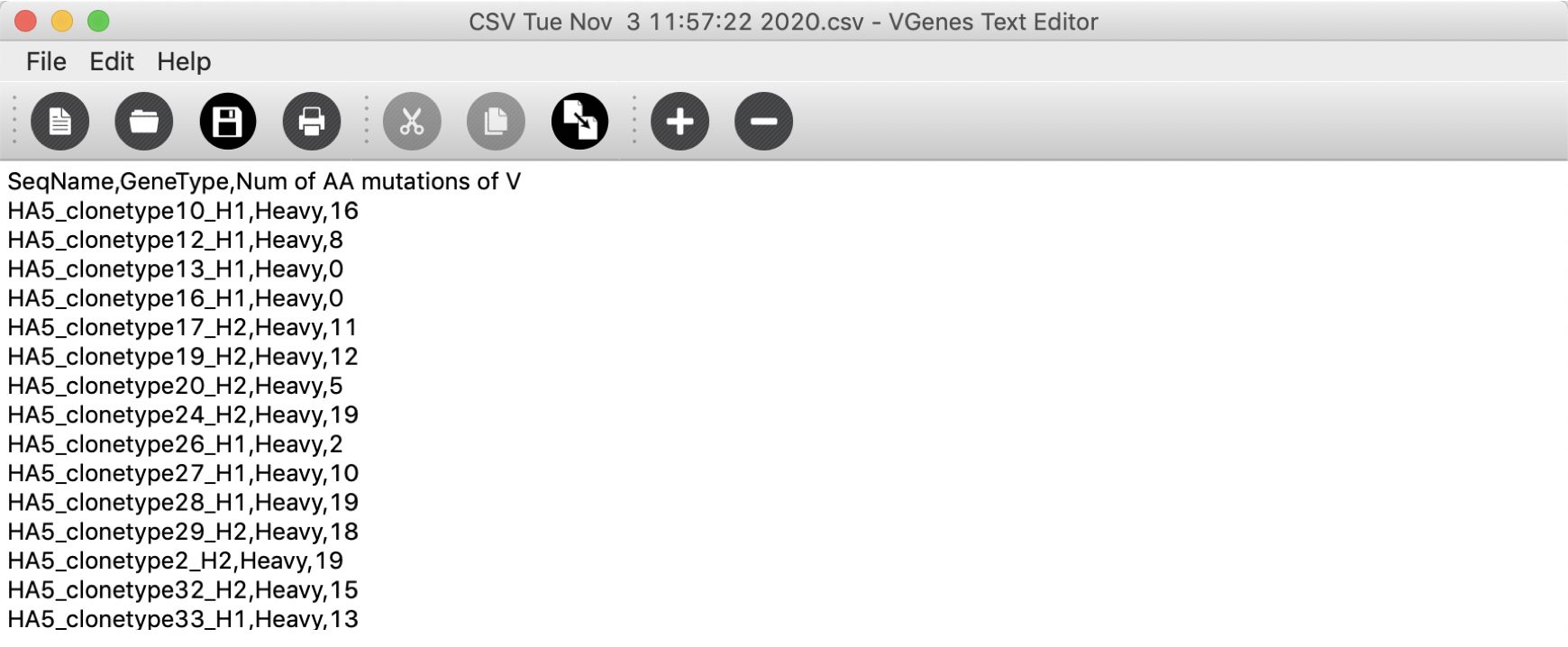
Amino Acid mutations report
This function will count and export number of amino acid mutations for selected sequences in CSV format.
-
Gibson Cloning sequence report
This function allows users to export their selected antibodies for Gibson cloning. VGenes developed a graphical UI to preview all selected sequences for users to confirm. VGenes automatically checks the end of sequences, and the ORF correction. All sequence passed checked will be highlighted in green with a “Good” indicator. Two error messages, “Jend error” and “ORF error”, are highlighted in red to attract users’ attention. Users are allowed to directly edit sequences in the text edit to fix the Jend error or ORF error. Indicators will be automatically updated when users edit the corresponding sequences. At last, VGenes allows users to make their own decision: users can click “I think this sequence is OK” button to compulsorily set indicator of a sequence to “Good” even to ignore all error messages. After users confirmed all sequences, users should provide Gibson upstream and downstream connector sequences. After users click “Confirm”, VGenes will generated sequences for all records with indicator “Good” in a CSV file.